
Decoding the Epigenome of Breast Cancer

 , , , and
, , , and
Abstract
:1. Breast Cancer
2. Epigenetics in BC
3. DNA Methylation in BC
3.1. Diagnostic Potential of DNA Methylation in BC Subtyping
3.2. Impact of DNA Methylome on Signaling Pathways and Prognostic Implications
3.3. DNA Methylation as a Regulator of Therapy Response
4. Histone Modifications in BC
4.1. Diagnostic Potential of Histone Modifications in BC Subtyping
4.2. Prognostic Implications of Histone Modifications

{kind=link}
{kind=link}
{kind=link}
| Enzyme | Substrate | Alteration | Association with Clinical Parameters | Refs | |
|---|---|---|---|---|---|
| HMTs | EZH2 | H3K27me1/2/3 | ↑ in invasive carcinoma and metastasis | - | [138,139] |
| DOT1L | H3K79me1/me2/me3 | ↑ in ER− BC | Poor survival and aggressiveness | [149,150] | |
| MLL2 (KMT2B) | H3K4me2/me3 | ↓ in BC | none | [151] | |
| MLL3 (KMT2C) | H3K4me1/2 | ↓ in hormone negative BC | - | [152] | |
| SUV4-20H2 (KMT5C) | H4K20me3 | ↓ in BC | - | [120] | |
| SETD1A | H3K4me | ↑ in BC | - | [153,154] | |
| NSD3 | H3K36me2/me3 | ↑ in BC | Worse overall and disease-free survival | [155] | |
| G9a (EHMT2) | H3K9me1/me2 | ↑ in BC | Poor outcome | [156] | |
| HDMTs | KDM1A | H3K4me1/2 H3K9me1/2 | ↑ with DCIS and IDC advancement ↑ in basal-like BC | - Poor outcome | [157,158] |
| KDM2B | H3K4me3 H3K36me2/3 | ↑ in TNBC | Poor prognosis/early relapse | [159,160] | |
| KDM3A | H3K9me1/2 | ↑ in BC | - | [135] | |
| KDM4A/B/C | H3K9me3, H3K36me2/3 | ↑ in ER+ BC | - | [141,142] | |
| KDM5A | H3K4me2/3 | ↑ in BC | Therapy resistance | [161] | |
| KDM5B | H3K4me1/2/3 | ↑ in HER2+ BC ↓ in basal-like BC | Poor outcome in ER+ BC | [162] | |
| KDM6B | H3K27me2/3 | ↓ in BC | Poor prognosis | [163] | |
| HATs | p300 | H3 (K14, K18, K23) H4 (K5, K8, K12) | ↑ in BC | Grade, clinical stage, and tumor size, and recurrence | [143,164,165] |
| CBP | H3 (K14, K18, K23) H4 (K5, K8, K12) | ↑ in luminal A and B BC | ER and PR expression | [166] | |
| KAT2A | H3 (K9, K14, K18, K23) | ↑ in BC | Tamoxifen-resistance | [166,167] | |
| KAT5 | H2A, H3, H4 | ↓ in BC | - | [168] | |
| KAT6A | H3 (K9, K14) H4 (K5, K8, K12) | ↑ in BC | ERα expression Worse clinical outcome | [169] | |
| KAT7 | H4 (K5, K8, K12, K16) | ↑ in BC | Worse clinical outcome | [144] | |
| HDACs | HDAC1 | H3, H4 | ↑ in BC | ER/HER2 expression | [146,170] |
| HDAC2 | H3K56, H4K16 | ↑ in poorly differentiated BC | HER2 status | [147] | |
| HDAC3 | H3K9ac | ↑ in poorly differentiated BC | ER/HER2 expression | [148] | |
| HDAC5 | H3 (K9, K14) H4 (K5, K8, K12) | ↑ in BC | - | [171] | |
| HDAC8 | H3, H4 | ↑ in BC | - | [172,173] | |
| HDAC9 | H3, H4 | ↑ in aggressive and tamoxifen-resistant BC | - | [174] | |
| HDAC11 | H2A, H2B, H3, H4 | ↓ in basal-like BC | Unfavorable prognosis | [175] | |
| SIRT1 | H3 (K9, K56) H4K16 | ↑ in BC | Tumor size and grade, lymph node, metastasis | [176] |
4.3. Therapeutic Implications of Histone Modifications in BC
5. Non-Coding RNAs in BC
5.1. Diagnostic and Prognostic Potential of ncRNAs
5.2. Therapeutic Potential of ncRNAs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Lüönd, F.; Tiede, S.; Christofori, G. Breast Cancer as An Example of Tumour Heterogeneity and Tumour Cell Plasticity during Malignant Progression. Br. J. Cancer 2021, 125, 164–175. [Google Scholar] [CrossRef]
- Rakha, E.; Toss, M.; Quinn, C. Specific Cell Differentiation in Breast Cancer: A Basis for Histological Classification. J. Clin. Pathol. 2022, 75, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Al-Benna, S.; Poggemann, K.; Steinau, H.U.; Steinstraesser, L. Diagnosis and Management of Primary Breast Sarcoma. Breast Cancer Res. Treat. 2010, 122, 619–626. [Google Scholar] [CrossRef]
- Alghodhaifi, H.; Alghodhaifi, A.; Alghodhaifi, M. Predicting Invasive Ductal Carcinoma in Breast Histology Images Using Convolutional Neural Network. In Proceedings of the 2019 IEEE National Aerospace and Electronics Conference (NAECON), Dayton, OH, USA, 15–19 July 2019; pp. 374–378. [Google Scholar]
- Sultan, G.; Zubair, S.; Tayubi, I.A.; Dahms, H.-U.; Madar, I.H. Towards the Early Detection of Ductal Carcinoma (a Common Type of Breast Cancer) Using Biomarkers Linked to the PPAR(γ) Signaling Pathway. Bioinformation 2019, 15, 799–805. [Google Scholar] [CrossRef]
- Lee, Y.M.; Oh, M.H.; Go, J.H.; Han, K.; Choi, S.Y. Molecular Subtypes of Triple-Negative Breast Cancer: Understanding of Subtype Categories and Clinical Implication. Genes Genom. 2020, 42, 1381–1387. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer; Exon Publications: Brisbane, Australia, 2022; pp. 31–42. [Google Scholar]
- Lee, A.; Moon, B.I.; Kim, T.H. BRCA1/BRCA2 Pathogenic Variant Breast Cancer: Treatment and Prevention Strategies. Ann. Lab. Med. 2020, 40, 114–121. [Google Scholar] [CrossRef]
- Breast Cancer Association Consortium. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- McDevitt, T.; Durkie, M.; Arnold, N.; Burghel, G.J.; Butler, S.; Claes, K.B.M.; Logan, P.; Robinson, R.; Sheils, K.; Wolstenholme, N.; et al. EMQN Best Practice Guidelines for Genetic Testing in Hereditary Breast and Ovarian Cancer. Eur. J. Hum. Genet. 2024, 32, 479–488. [Google Scholar] [CrossRef]
- Shahbandi, A.; Nguyen, H.D.; Jackson, J.G. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer 2020, 6, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Kechagioglou Papi, R.M.; Provatopoulou, X.; Kalogera, E.; Papadimitriou, E.; Grigoropoulos, P.; Nonni, A.; Zografos, G.; Kyriakidis, D.A.; Gounaris, A.P. Tumor Suppressor PTEN in Breast Cancer: Heterozygosity, Mutations and Protein Expression. Anticancer Res. 2014, 34, 1387–1400. [Google Scholar]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM Mutations That Cause Ataxia-Telangiectasia Are Breast Cancer Susceptibility Alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Meijers-Heijboer, H.; Van den Ouweland, A.; Klijn, J.; Wasielewski, M.; De Shoo, A.; Oldenburg, R.; Hollestelle, A.; Houben, M.; Crepin, E.; Van Veghel-Plandsoen, M.; et al. Low-Penetrance Susceptibility to Breast Cancer Due to CHEK2*1100delC in Noncarriers of BRCA1 or BRCA2 Mutations: The CHEK2-Breast Cancer Consortium. Nat. Genet. 2002, 31, 55–59. [Google Scholar] [CrossRef]
- Bose, M.; Benada, J.; Thatte, J.V.; Kinalis, S.; Ejlertsen, B.; Nielsen, F.C.; Sørensen, C.S.; Rossing, M. A Catalog of Curated Breast Cancer Genes. Breast Cancer Res. Treat. 2022, 191, 431–441. [Google Scholar] [CrossRef]
- Sarhangi, N.; Hajjari, S.; Heydari, S.F.; Ganjizadeh, M.; Rouhollah, F.; Hasanzad, M. Breast Cancer in the Era of Precision Medicine. Mol. Biol. Rep. 2022, 49, 10023–10037. [Google Scholar] [CrossRef]
- Riis, M. Modern Surgical Treatment of Breast Cancer. Ann. Med. Surg. 2020, 56, 95–107. [Google Scholar] [CrossRef]
- Korde, L.A.; Somerfield, M.R.; Carey, L.A.; Crews, J.R.; Denduluri, N.; Hwang, E.S.; Khan, S.A.; Loibl, S.; Morris, E.A.; Perez, A.; et al. Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline. J. Clin. Oncol. 2021, 39, 1485–1505. [Google Scholar] [CrossRef]
- Costa, B.; Amorim, I.; Gärtner, F.; Vale, N. Understanding Breast Cancer: From Conventional Therapies to Repurposed Drugs. Eur. J. Pharm. Sci. 2020, 151, 105401. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, S.; Xin, Q.; Zhang, Y.; Wang, K.; Li, M. Recent Progress of CDK4/6 Inhibitors’ Current Practice in Breast Cancer. Cancer Gene Ther. 2024, 31, 1283–1291. [Google Scholar] [CrossRef]
- Ye, F.; Dewanjee, S.; Li, Y.; Jha, N.K.; Chen, Z.S.; Kumar, A.; Vishakha; Behl, T.; Jha, S.K.; Tang, H. Advancements in Clinical Aspects of Targeted Therapy and Immunotherapy in Breast Cancer. Mol. Cancer 2023, 22, 105. [Google Scholar] [CrossRef] [PubMed]
- Schlam, I.; Swain, S.M. HER2-Positive Breast Cancer and Tyrosine Kinase Inhibitors: The Time Is Now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987. [Google Scholar] [CrossRef]
- Li, L.; Zhang, F.; Liu, Z.; Fan, Z. Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome. Cancers 2023, 15, 321. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 Pathway: Basic Biology and Role in Cancer Immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Bombonati, A.; Sgroi, D.C. The Molecular Pathology of Breast Cancer Progression. J. Pathol. 2011, 223, 308–318. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Ct, W.; Morris, J.R. Genes, Genetics, and Epigenetics: A Correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef]
- Recillas-Targa, F. Cancer Epigenetics: An Overview. Arch. Med. Res. 2022, 53, 732–740. [Google Scholar] [CrossRef]
- El Helou, R.; Wicinski, J.; Guille, A.; Adélaïde, J.; Finetti, P.; Bertucci, F.; Chaffanet, M.; Birnbaum, D.; Charafe-Jauffret, E.; Ginestier, C. Brief Reports: A Distinct DNA Methylation Signature Defines Breast Cancer Stem Cells and Predicts Cancer Outcome. Stem Cells 2014, 32, 3031–3036. [Google Scholar] [CrossRef]
- Bloushtain-Qimron, N.; Yao, J.; Snyder, E.L.; Shipitsin, M.; Campbell, L.L.; Mani, S.A.; Hu, M.; Chen, H.; Ustyansky, V.; Antosiewicz, J.E.; et al. Cell Type-Specific DNA Methylation Patterns in the Human Breast. Proc. Natl. Acad. Sci. USA 2008, 105, 14076–14081. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA Methylation in Breast Cancer Identifies Novel Prognostic Biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Sousa, M.; Amorim, M.; Salta, S.; De Sousa, S.P.; Henrique, R.; Jerónimo, C. Predicting Resistance to Endocrine Therapy in Breast Cancer: It’s Time for Epigenetic Biomarkers (Review). Oncol. Rep. 2019, 41, 1431–1438. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Wang, C.; Wang, X. TET (Ten-Eleven Translocation) Family Proteins: Structure, Biological Functions and Applications. Signal Transduct. Target. Ther. 2023, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Loaeza-Loaeza, J.; Beltran, A.S.; Hernández-Sotelo, D. DNMTs and Impact of CpG Content, Transcription Factors, Consensus Motifs, LncRNAs, and Histone Marks on DNA Methylation. Genes 2020, 11, 1336. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Zeng, F.; Li, W.; He, Z.; Chen, W.; Zhu, W.; Zhang, B. The Prognostic Value of Global DNA Hypomethylation in Cancer: A Meta-Analysis. PLoS ONE 2014, 9, e106290. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA Hypermethylation in Disease: Mechanisms and Clinical Relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA Hypomethylation Coupled to Repressive Chromatin Domain Formation and Gene Silencing in Breast Cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef]
- Lehmann, U.; Länger, F.; Feist, H.; Glöckner, S.; Hasemeier, B.; Kreipe, H. Quantitative Assessment of Promoter Hypermethylation during Breast Cancer Development. Am. J. Pathol. 2002, 160, 605–612. [Google Scholar] [CrossRef]
- Jackson, K.; Yu, M.C.; Arakawa, K.; Fiala, E.; Youn, B.; Fiegl, H.; Müller-Holzner, E.; Widschwendter, M.; Ehrlich, M. DNA Hypomethylation Is Prevalent Even in Low-Grade Breast Cancers. Cancer Biol. Ther. 2004, 3, 1225–1231. [Google Scholar] [CrossRef]
- Lindqvist, B.M.; Wingren, S.; Motlagh, P.B.; Nilsson, T.K. Whole Genome DNA Methylation Signature of HER2-Positive Breast Cancer. Epigenetics 2014, 9, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Zarean, E.; Li, S.; Wong, E.M.; Makalic, E.; Milne, R.L.; Giles, G.G.; McLean, C.; Southey, M.C.; Dugué, P.-A. Tumour DNA Methylation Markers Associated with Breast Cancer Survival: A Replication Study. Breast Cancer Res. 2025, 27, 9. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Grushko, T.A.; Dignam, J.; Hagos, F.; Nanda, R.; Sveen, L.; Xu, J.; Fackenthal, J.; Tretiakova, M.; Das, S.; et al. BRCA1 Promoter Methylation in Sporadic Breast Cancer Is Associated with Reduced BRCA1 Copy Number and Chromosome 17 Aneusomy. Cancer Res. 2005, 65, 10692–10699. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wei, W.; Jiang, Y.; Yang, H.; Liu, J. Promoter Methylation and Expression Changes of BRCA1 in Cancerous Tissues of Patients with Sporadic Breast Cancer. Oncol. Lett. 2015, 9, 1807–1813. [Google Scholar] [CrossRef]
- Jacot, W.; Lopez-Crapez, E.; Mollevi, C.; Boissière-Michot, F.; Simony-Lafontaine, J.; Ho-Pun-Cheung, A.; Chartron, E.; Theillet, C.; Lemoine, A.; Saffroy, R.; et al. BRCA1 Promoter Hypermethylation Is Associated with Good Prognosis and Chemosensitivity in Triple-Negative Breast Cancer. Cancers 2020, 12, 828. [Google Scholar] [CrossRef]
- Oubaddou, Y.; Oukabli, M.; Fenniche, S.; Elktaibi, A.; Elochi, M.R.; Al Bouzidi, A.; Qmichou, Z.; Dakka, N.; Diorio, C.; Richter, A.; et al. BRCA1 Promoter Hypermethylation in Malignant Breast Tumors and in the Histologically Normal Adjacent Tissues to the Tumors: Exploring Its Potential as a Biomarker and Its Clinical Significance in a Translational Approach. Genes 2023, 14, 1680. [Google Scholar] [CrossRef]
- Asiaf, A.; Ahmad, S.T.; Malik, A.A.; Aziz, S.A.; Rasool, Z.; Masood, A.; Zargar, M.A. Protein Expression and Methylation of MGMT, a DNA Repair Gene and Their Correlation with Clinicopathological Parameters in Invasive Ductal Carcinoma of the Breast. Tumor Biol. 2015, 36, 6485–6496. [Google Scholar] [CrossRef]
- Fumagalli, C.; Pruneri, G.; Possanzini, P.; Manzotti, M.; Barile, M.; Feroce, I.; Colleoni, M.; Bonanni, B.; Maisonneuve, P.; Radice, P.; et al. Methylation of O 6-Methylguanine-DNA Methyltransferase (MGMT) Promoter Gene in Triple-Negative Breast Cancer Patients. Breast Cancer Res. Treat. 2012, 134, 131–137. [Google Scholar] [CrossRef]
- Shargh, S.A.; Sakizli, M.; Khalaj, V.; Movafagh, A.; Yazdi, H.; Hagigatjou, E.; Sayad, A.; Mansouri, N.; Mortazavi-Tabatabaei, S.A.; Khorram Khorshid, H.R. Downregulation of E-Cadherin Expression in Breast Cancer by Promoter Hypermethylation and Its Relation with Progression and Prognosis of Tumor. Med. Oncol. 2014, 31, 250. [Google Scholar] [CrossRef]
- Liu, J.; Sun, X.; Qin, S.; Wang, H.; Du, N.; Li, Y.; Pang, Y.; Wang, C.; Xu, C.; Ren, H. CDH1 Promoter Methylation Correlates with Decreased Gene Expression and Poor Prognosis in Patients with Breast Cancer. Oncol. Lett. 2016, 11, 2635–2643. [Google Scholar] [CrossRef]
- Yari, K.; Rahimi, Z. Promoter Methylation Status of the Retinoic Acid Receptor-Beta 2 Gene in Breast Cancer Patients: A Case Control Study and Systematic Review. Breast Care 2019, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Van Hoesel, A.Q.; Sato, Y.; Elashoff, D.A.; Turner, R.R.; Giuliano, A.E.; Shamonki, J.M.; Kuppen, P.J.K.; Van De Velde, C.J.H.; Hoon, D.S.B. Assessment of DNA Methylation Status in Early Stages of Breast Cancer Development. Br. J. Cancer 2013, 108, 2033–2038. [Google Scholar] [CrossRef] [PubMed]
- Swellam, M.; Abdelmaksoud, M.D.E.; Sayed Mahmoud, M.; Ramadan, A.; Abdel-Moneem, W.; Hefny, M.M. Aberrant Methylation of APC and RARβ2genes in Breast Cancer Patients. IUBMB Life 2015, 67, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-S.; Wang, S.-C.; Yen, Y.-T.; Lee, T.-H.; Wen, W.-C.; Lin, R.-K. Hypermethylation of CCND2 in Lung and Breast Cancer Is a Potential Biomarker and Drug Target. Int. J. Mol. Sci. 2018, 19, 3096. [Google Scholar] [CrossRef]
- Kajabova, V.; Smolkova, B.; Zmetakova, I.; Sebova, K.; Krivulcik, T.; Bella, V.; Kajo, K.; Machalekova, K.; Fridrichova, I. RASSF1A Promoter Methylation Levels Positively Correlate with Estrogen Receptor Expression in Breast Cancer Patients. Transl. Oncol. 2013, 6, 297-IN5. [Google Scholar] [CrossRef]
- Silva, J.; Domínguez, G.; Silva, J.M.; GarcõÂ, J.M.; Gallego, I.; Corbacho, C.; Provencio, M.; EspanÄ, P.; Bonilla, F. Analysis of Genetic and Epigenetic Processes That Influence P14ARF Expression in Breast Cancer. Oncogene 2001, 20, 4586–4790. [Google Scholar] [CrossRef]
- Zhang, S.L.; Wang, Y.Q.; Zhang, J.H.; Hu, J.W.; Ma, J.; Gu, Z.; Wang, Y.; Chen, J.J. Methylated P16 Gene Is Associated with Negative Expression of Estrogen Receptor, Progesterone Receptor and Human Epidermal Growth Factor Receptor 2 in Breast Cancer. Eur. J. Gynaecol. Oncol. 2021, 42, 530. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, C.; Jiang, H.; Liang, L.; Shi, W.; Zhang, Q.; Sun, P.; Xiang, R.; Wang, Y.; Yang, S. ZEB1 Induces ER-α Promoter Hypermethylation and Confers Antiestrogen Resistance in Breast Cancer. Cell Death Dis. 2017, 8, e2732. [Google Scholar] [CrossRef]
- Intabli, H.; Gee, J.M.; Oesterreich, S.; Yeoman, M.S.; Allen, M.C.; Qattan, A.; Flint, M.S. Glucocorticoid Induced Loss of Oestrogen Receptor Alpha Gene Methylation and Restoration of Sensitivity to Fulvestrant in Triple Negative Breast Cancer. Gene 2023, 851, 147022. [Google Scholar] [CrossRef]
- García, J.M.; Silva, J.; Peña, C.; Garcia, V.; Rodríguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; España, P.; Bonilla, F. Promoter Methylation of the PTEN Gene is a Common Molecular Change in Breast Cancer. Genes Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef]
- Fan, Y.; Xie, G.; Wang, Z.; Wang, Y.; Wang, Y.; Zheng, H.; Zhong, X. PTEN Promoter Methylation Predicts 10-Year Prognosis in Hormone Receptor-Positive Early Breast Cancer Patients Who Received Adjuvant Tamoxifen Endocrine Therapy. Breast Cancer Res. Treat. 2022, 192, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, A.; Hashim, M.; Abouzid, A.; Swellam, M. Clinical Impact of PTEN Methylation Status as a Prognostic Marker for Breast Cancer. J. Genet. Eng. Biotechnol. 2021, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Saelee, P.; Pongtheerat, T. APC Promoter Hypermethylation as a Prognostic Marker in Breast Cancer Patients. Asian Pac. J. Cancer Prev. 2020, 21, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Van Hoesel, A.Q.; Van De Velde, C.J.H.; Kuppen, P.J.K.; Liefers, G.J.; Putter, H.; Sato, Y.; Elashoff, D.A.; Turner, R.R.; Shamonki, J.M.; De Kruijf, E.M.; et al. Hypomethylation of LINE-1 in Primary Tumor Has Poor Prognosis in Young Breast Cancer Patients: A Retrospective Cohort Study. Breast Cancer Res. Treat. 2012, 134, 1103–1114. [Google Scholar] [CrossRef]
- Ramos, E.A.S.; Grochoski, M.; Braun-Prado, K.; Seniski, G.G.; Cavalli, I.J.; Ribeiro, E.M.S.F.; Camargo, A.A.; Costa, F.F.; Klassen, G. Epigenetic Changes of CXCR4 and Its Ligand CXCL12 as Prognostic Factors for Sporadic Breast Cancer. PLoS ONE 2011, 6, e29461. [Google Scholar] [CrossRef]
- Li, S.Y.; Rong, M.; Iacopetta, B. DNA Hypermethylation in Breast Cancer and Its Association with Clinicopathological Features. Cancer Lett. 2006, 237, 272–280. [Google Scholar] [CrossRef]
- Győrffy, B.; Bottai, G.; Fleischer, T.; Munkácsy, G.; Budczies, J.; Paladini, L.; Børresen-Dale, A.-L.; Kristensen, V.N.; Santarpia, L. Aberrant DNA Methylation Impacts Gene Expression and Prognosis in Breast Cancer Subtypes. Int. J. Cancer 2016, 138, 87–97. [Google Scholar] [CrossRef]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallon-Christersson, J.; Jönsson, G.; Olsson, H.; Borg, Å.; Ringnér, M. Molecular Subtypes of Breast Cancer Are Associated with Characteristic DNA Methylation Patterns. Breast Cancer Res. 2010, 12, R36. [Google Scholar] [CrossRef]
- Stefansson, O.A.; Moran, S.; Gomez, A.; Sayols, S.; Arribas-Jorba, C.; Sandoval, J.; Hilmarsdottir, H.; Olafsdottir, E.; Tryggvadottir, L.; Jonasson, J.G.; et al. A DNA Methylation-based Definition of Biologically Distinct Breast Cancer Subtypes. Mol. Oncol. 2015, 9, 555–568. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bièche, I. Expression Analysis of DNA Methyltransferases 1, 3A, and 3B in Sporadic Breast Carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar]
- Wong, K.K. DNMT1: A Key Drug Target in Triple-Negative Breast Cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Roll, J.D.; Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNMT3b Overexpression Contributes to a Hypermethylator Phenotype in Human Breast Cancer Cell Lines. Mol. Cancer 2008, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, T.; Frigessi, A.; Johnson, K.C.; Edvardsen, H.; Touleimat, N.; Klajic, J.; Riis, M.L.; Haakensen, V.D.; Wärnberg, F.; Naume, B.; et al. Genome-Wide DNA Methylation Profiles in Progression to in Situand Invasive Carcinoma of the Breast with Impact on Gene Transcription and Prognosis. Genome Biol. 2014, 15, 435. [Google Scholar] [CrossRef]
- Johnson, K.C.; Koestler, D.C.; Fleischer, T.; Chen, P.; Jenson, E.G.; Marotti, J.D.; Onega, T.; Kristensen, V.N.; Christensen, B.C. DNA Methylation in Ductal Carcinoma In Situ Related with Future Development of Invasive Breast Cancer. Clin. Epigenetics 2015, 7, 75. [Google Scholar] [CrossRef]
- Raos, D.; Ulamec, M.; Bojanac, A.K.; Bulic-Jakus, F.; Jezek, D.; Sincic, N. Epigenetically Inactivated RASSF1A as a Tumor Biomarker. Bosn. J. Basic Med. Sci. 2021, 21, 386–397. [Google Scholar] [CrossRef]
- Dubois, F.; Bergot, E.; Zalcman, G.; Levallet, G. RASSF1A, Puppeteer of Cellular Homeostasis, Fights Tumorigenesis, and Metastasis—An Updated Review. Cell Death Dis. 2019, 10, 928. [Google Scholar] [CrossRef]
- Ruscito, I.; Gasparri, M.L.; De Marco, M.P.; Costanzi, F.; Besharat, A.R.; Papadia, A.; Kuehn, T.; Gentilini, O.D.; Bellati, F.; Caserta, D. The Clinical and Pathological Profile of BRCA1 Gene Methylated Breast Cancer Women: A Meta-Analysis. Cancers 2021, 13, 1391. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; El Salhat, H.; Taha, R.Z.; John, A.; Ali, B.R.; Elkord, E. DNA Methylation and Repressive H3K9 and H3K27 Trimethylation in the Promoter Regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 Genes in Human Primary Breast Cancer. Clin. Epigenetics 2018, 10, 78. [Google Scholar] [CrossRef]
- Man, X.; Li, Q.; Wang, B.; Zhang, H.; Zhang, S.; Li, Z. DNMT3A and DNMT3B in Breast Tumorigenesis and Potential Therapy. Front. Cell Dev. Biol. 2022, 10, 916725. [Google Scholar] [CrossRef]
- Kar, S.; Sengupta, D.; Deb, M.; Shilpi, A.; Parbin, S.; Rath, S.K.; Pradhan, N.; Rakshit, M.; Patra, S.K. Expression Profiling of DNA Methylation-Mediated Epigenetic Gene-Silencing Factors in Breast Cancer. Clin. Epigenetics 2014, 6, 20. [Google Scholar] [CrossRef]
- Yu, Z.; Xiao, Q.; Zhao, L.; Ren, J.; Bai, X.; Sun, M.; Wu, H.; Liu, X.; Song, Z.; Yan, Y.; et al. DNA Methyltransferase 1/3a Overexpression in Sporadic Breast Cancer Is Associated with Reduced Expression of Estrogen Receptor-Alpha/Breast Cancer Susceptibility Gene 1 and Poor Prognosis. Mol. Carcinog. 2015, 54, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Rodenhiser, D.I. Epigenetic Inactivation of BRCA1 Is Associated with Aberrant Expression of CTCF and DNA Methyltransferase (DNMT3B) in Some Sporadic Breast Tumours. Eur. J. Cancer 2007, 43, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chang, Z.; Shi, K.; Song, L.; Cui, L.; Ma, Z.; Li, X.; Ma, W.; Wang, L. The Correlation between DNMT1 and ERα Expression and the Methylation Status of ERα, and Its Clinical Significance in Breast Cancer. Oncol. Lett. 2016, 11, 1995–2000. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef]
- Palomeras, S.; Diaz-Lagares, Á.; Viñas, G.; Setien, F.; Ferreira, H.J.; Oliveras, G.; Crujeiras, A.B.; Hernández, A.; Lum, D.H.; Welm, A.L.; et al. Epigenetic Silencing of TGFBI Confers Resistance to Trastuzumab in Human Breast Cancer. Breast Cancer Res. 2019, 21, 79. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Mahalingaiah, P.K.S.; Chang, Y.-W.; Singh, K.P. Reversal of Epigenetic Aberrations Associated with the Acquisition of Doxorubicin Resistance Restores Drug Sensitivity in Breast Cancer Cells. Eur. J. Pharm. Sci. 2018, 123, 56–69. [Google Scholar] [CrossRef]
- Chekhun, V.F.; Kulik, G.I.; Yurchenko, O.V.; Tryndyak, V.P.; Todor, I.N.; Luniv, L.S.; Tregubova, N.A.; Pryzimirska, T.V.; Montgomery, B.; Rusetskaya, N.V.; et al. Role of DNA Hypomethylation in the Development of the Resistance to Doxorubicin in Human MCF-7 Breast Adenocarcinoma Cells. Cancer Lett. 2006, 231, 87–93. [Google Scholar] [CrossRef]
- Widschwendter, M.; Siegmund, K.D.; Müller, H.M.; Fiegl, H.; Marth, C.; Müller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of Breast Cancer DNA Methylation Profiles with Hormone Receptor Status and Response to Tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Klajic, J.; Busato, F.; Edvardsen, H.; Touleimat, N.; Fleischer, T.; Bukholm, I.; Børresen-Dale, A.-L.; Lønning, P.E.; Tost, J.; Kristensen, V.N. DNA Methylation Status of Key Cell-Cycle Regulators Such as CDKNA2/P16 and CCNA1 Correlates with Treatment Response to Doxorubicin and 5-Fluorouracil in Locally Advanced Breast Tumors. Clin. Cancer Res. 2014, 20, 6357–6366. [Google Scholar] [CrossRef]
- Pedersen, C.A.; Cao, M.D.; Fleischer, T.; Rye, M.B.; Knappskog, S.; Eikesdal, H.P.; Lønning, P.E.; Tost, J.; Kristensen, V.N.; Tessem, M.-B.; et al. DNA Methylation Changes in Response to Neoadjuvant Chemotherapy Are Associated with Breast Cancer Survival. Breast Cancer Res. 2022, 24, 43. [Google Scholar] [CrossRef]
- Bárcena-Varela, M.; Caruso, S.; Llerena, S.; Álvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef]
- Li, Z.; Li, B.; Yu, H.; Wang, P.; Wang, W.; Hou, P.; Li, M.; Chu, S.; Zheng, J.; Mao, L.; et al. DNMT1-Mediated Epigenetic Silencing of TRAF6 Promotes Prostate Cancer Tumorigenesis and Metastasis by Enhancing EZH2 Stability. Oncogene 2022, 41, 3991–4002. [Google Scholar] [CrossRef]
- Xing, J.; Stewart, D.J.; Gu, J.; Lu, C.; Spitz, M.R.; Wu, X. Expression of Methylation-Related Genes Is Associated with Overall Survival in Patients with Non-Small Cell Lung Cancer. Br. J. Cancer 2008, 98, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.; Sprowls, S.; Szalai, G.; Arsiwala, T.; Saralkar, P.; Straight, B.; Hatcher, S.; Tyree, E.; Yost, M.; Kohler, W.J.; et al. Hypomethylating Agent Azacitidine Is Effective in Treating Brain Metastasis Triple-Negative Breast Cancer Through Regulation of DNA Methylation of Keratin 18 Gene. Transl. Oncol. 2020, 13, 100775. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, R.; Mosaffa, F.; Emami Razavi, A.; Teimoori-Toolabi, L.; Jamialahmadi, K. Altered DNA Methyltransferases Promoter Methylation and MRNA Expression Are Associated with Tamoxifen Response in Breast Tumors. J. Cell. Physiol. 2018, 233, 7305–7319. [Google Scholar] [CrossRef] [PubMed]
- Dahn, M.L.; Cruickshank, B.M.; Jackson, A.J.; Dean, C.; Holloway, R.W.; Hall, S.R.; Coyle, K.M.; Maillet, H.; Waisman, D.M.; Goralski, K.B.; et al. Decitabine Response in Breast Cancer Requires Efficient Drug Processing and Is Not Limited by Multidrug Resistance. Mol. Cancer Ther. 2020, 19, 1110–1122. [Google Scholar] [CrossRef]
- Khan, G.N.; Kim, E.J.; Shin, T.S.; Lee, S.H. Azacytidine-Induced Chemosensitivity to Doxorubicin in Human Breast Cancer MCF7 Cells. Anticancer Res. 2017, 37, 2355–2364. [Google Scholar] [CrossRef]
- Connolly, R.M.; Li, H.; Jankowitz, R.C.; Zhang, Z.; Rudek, M.A.; Jeter, S.C.; Slater, S.A.; Powers, P.; Wolff, A.C.; Fetting, J.H.; et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin. Cancer Res. 2017, 23, 2691–2701. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone Modifications in Epigenetic Regulation of Cancer: Perspectives and Achieved Progress. Semin. Cancer Biol. 2022, 83, 452–471. [Google Scholar] [CrossRef]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation and Co: An Expanding Repertoire of Histone Acylations Regulates Chromatin and Transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.E.; Venkatasubramani, A.V.; Pathirana, D.; Krause, S.; Sparr, A.C.; Hasenauer, J.; Imhof, A.; Müller, M.; Becker, P.B. Processivity and Specificity of Histone Acetylation by the Male-Specific Lethal Complex. Nucleic Acids Res. 2024, 52, 4889–4905. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Déjardin, J. The Chromatin Signatures of Enhancers and Their Dynamic Regulation. Nucleus 2023, 14, 2160551. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The Diverse Functions of Histone Lysine Methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Gates, L.A.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’: Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef]
- Yoo, K.H.; Hennighausen, L. EZH2 Methyltransferase and H3K27 Methylation in Breast Cancer. Int. J. Biol. Sci. 2012, 8, 59–65. [Google Scholar] [CrossRef]
- Alseksek, R.K.; Ramadan, W.S.; Saleh, E.; El-Awady, R. The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 8141. [Google Scholar] [CrossRef]
- Liang, T.; Wang, F.; Elhassan, R.M.; Cheng, Y.; Tang, X.; Chen, W.; Fang, H.; Hou, X. Targeting Histone Deacetylases for Cancer Therapy: Trends and Challenges. Acta Pharm. Sin. B 2023, 13, 2425–2463. [Google Scholar] [CrossRef]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-K.; Ren, Y.; Chen, Q.; Yang, Y.; Tang, L.; Xu, L.; Ren, Z. H4K20me3, H3K4me2 and H3K9me2 Mediate the Effect of ER on Prognosis in Breast Cancer. Epigenetics 2024, 19, 2343593. [Google Scholar] [CrossRef] [PubMed]
- Mungamuri, S.K.; Murk, W.; Grumolato, L.; Bernstein, E.; Aaronson, S.A. Chromatin Modifications Sequentially Enhance ErbB2 Expression in ErbB2-Positive Breast Cancers. Cell Rep. 2013, 5, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yan, J.; Chen, Q.; Yang, Y.; Li, Y.; Ren, Y.; Weng, Z.; Zhang, X.; Guan, J.; Tang, L.; et al. Association of H3K9me3 with Breast Cancer Prognosis by Estrogen Receptor Status. Clin. Epigenetics 2022, 14, 135. [Google Scholar] [CrossRef]
- Holm, K.; Grabau, D.; Lövgren, K.; Aradottir, S.; Gruvberger-Saal, S.; Howlin, J.; Saal, L.H.; Ethier, S.P.; Bendahl, P.-O.; Stål, O.; et al. Global H3K27 Trimethylation and EZH2 Abundance in Breast Tumor Subtypes. Mol. Oncol. 2012, 6, 494–506. [Google Scholar] [CrossRef]
- Healey, M.A.; Hu, R.; Beck, A.H.; Collins, L.C.; Schnitt, S.J.; Tamimi, R.M.; Hazra, A. Association of H3K9me3 and H3K27me3 Repressive Histone Marks with Breast Cancer Subtypes in the Nurses’ Health Study. Breast Cancer Res. Treat. 2014, 147, 639–651. [Google Scholar] [CrossRef]
- Judes, G.; Dagdemir, A.; Karsli-Ceppioglu, S.; Lebert, A.; Echegut, M.; Ngollo, M.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. H3K4 Acetylation, H3K9 Acetylation and H3K27 Methylation in Breast Tumor Molecular Subtypes. Epigenomics 2016, 8, 909–924. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Matsumoto, A.; Hieda, M.; Shinchi, Y.; Ogihara, E.; Hamada, M.; Nishioka, Y.; Kimura, H.; Yoshidome, K.; Tsujimoto, M.; et al. Loss of Histone H4K20 Trimethylation Predicts Poor Prognosis in Breast Cancer and Is Associated with Invasive Activity. Breast Cancer Res. 2014, 16, R66. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, M.; Gan, X.; Ren, Y.; Yang, Y.; Weng, Z.; Zhang, X.; Guan, J.; Tang, L.; Ren, Z. Combined Low Levels of H4K16ac and H4K20me3 Predicts Poor Prognosis in Breast Cancer. Int. J. Clin. Oncol. 2023, 28, 1147–1157. [Google Scholar] [CrossRef]
- Chen, X.; Hu, H.; He, L.; Yu, X.; Liu, X.; Zhong, R.; Shu, M. A Novel Subtype Classification and Risk of Breast Cancer by Histone Modification Profiling. Breast Cancer Res. Treat. 2016, 157, 267–279. [Google Scholar] [CrossRef]
- Marsolier, J.; Prompsy, P.; Durand, A.; Lyne, A.-M.; Landragin, C.; Trouchet, A.; Bento, S.T.; Eisele, A.; Foulon, S.; Baudre, L.; et al. H3K27me3 Conditions Chemotolerance in Triple-Negative Breast Cancer. Nat. Genet. 2022, 54, 459–468. [Google Scholar] [CrossRef]
- Liu, Q.; Kulak, M.V.; Borcherding, N.; Maina, P.K.; Zhang, W.; Weigel, R.J.; Qi, H.H. A novel HER2 gene body enhancer contributes to HER2 expression. Oncogene 2018, 37, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Dreijerink, K.M.; Groner, A.C.; Cheng, H.; Ohlson, C.E.; Reyes, J.; Lin, C.Y.; Bradner, J.; Zhao, J.J.; Roberts, T.M.; et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell. Rep. 2016, 15, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Berger, L.; Kolben, T.; Meister, S.; Kolben, T.M.; Schmoeckel, E.; Mayr, D.; Mahner, S.; Jeschke, U.; Ditsch, N.; Beyer, S. Expression of H3K4me3 and H3K9ac in Breast Cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Lebert, A.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. The Epigenetic Landscape of Promoter Genome-Wide Analysis in Breast Cancer. Sci. Rep. 2017, 7, 6597. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone Onco-Modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar] [CrossRef]
- Pfister, S.; Rea, S.; Taipale, M.; Mendrzyk, F.; Straub, B.; Ittrich, C.; Thuerigen, O.; Sinn, H.P.; Akhtar, A.; Lichter, P. The Histone Acetyltransferase HMOF Is Frequently Downregulated in Primary Breast Carcinoma and Medulloblastoma and Constitutes a Biomarker for Clinical Outcome in Medulloblastoma. Int. J. Cancer 2008, 122, 1207–1213. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.-M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Ruan, K.; Yamamoto, T.G.; Asakawa, H.; Chikashige, Y.; Kimura, H.; Masukata, H.; Haraguchi, T.; Hiraoka, Y. Histone H4 Acetylation Required for Chromatin Decompaction during DNA Replication. Sci. Rep. 2015, 5, 12720. [Google Scholar] [CrossRef]
- González-Bermúdez, L.; Genescà, A.; Terradas, M.; Martín, M. Role of H4K16 Acetylation in 53BP1 Recruitment to Double-Strand Break Sites in in Vitro Aged Cells. Biogerontology 2022, 23, 499–514. [Google Scholar] [CrossRef]
- Nowsheen, S.; Aziz, K.; Tran, P.T.; Gorgoulis, V.G.; Yang, E.S.; Georgakilas, A.G. Epigenetic Inactivation of DNA Repair in Breast Cancer. Cancer Lett. 2014, 342, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Kapoor-Vazirani, P.; Kagey, J.D.; Powell, D.R.; Vertino, P.M. Role of HMOF-Dependent Histone H4 Lysine 16 Acetylation in the Maintenance of TMS1/ASC Gene Activity. Cancer Res. 2008, 68, 6810–6821. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-Y.; Lei, P.-J.; Zhang, X.; Zheng, J.-Y.; Wang, H.-Y.; Zhao, J.; Li, Y.-M.; Ye, M.; Li, L.; Wei, G.; et al. Global Histone Modification Profiling Reveals the Epigenomic Dynamics during Malignant Transformation in a Four-Stage Breast Cancer Model. Clin. Epigenetics 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-L.; Lei, P.-J.; Zhao, Q.-Y.; Li, L.; Wei, G.; Wu, M. Epigenomic Analysis in a Cell-Based Model Reveals the Roles of H3K9me3 in Breast Cancer Transformation. Epigenomics 2017, 9, 1077–1092. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA Oxidation as Triggered by H3K9me2 Demethylation Drives Estrogen-Induced Gene Expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- López, C.; Barnon, M.T.; Beacon, T.H.; Nardocci, G.; Davie, J.R. The Key Role of Differential Broad H3K4me3 and H3K4ac Domains in Breast Cancer. Gene 2022, 826, 146463. [Google Scholar] [CrossRef]
- Adibfar, S.; Elveny, M.; Kashikova, H.S.; Mikhailova, M.V.; Farhangnia, P.; Vakili-Samiani, S.; Tarokhian, H.; Jadidi-Niaragh, F. The Molecular Mechanisms and Therapeutic Potential of EZH2 in Breast Cancer. Life Sci. 2021, 286, 120047. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q. The Roles of EZH2 in Cancer and Its Inhibitors. Med. Oncol. 2023, 40, 167. [Google Scholar] [CrossRef]
- Pei, J.; Zhang, S.Q.; Yang, X.; Han, C.; Pan, Y.; Li, J.; Wang, Z.; Sun, C.; Zhang, J. Epigenetic Regulator KDM4A Activates Notch1-NICD-Dependent Signaling to Drive Tumorigenesis and Metastasis in Breast Cancer. Transl. Oncol. 2023, 28, 101615. [Google Scholar] [CrossRef]
- Gaughan, L.; Stockley, J.; Coffey, K.; O’Neill, D.; Jones, D.L.; Wade, M.; Wright, J.; Moore, M.; Tse, S.; Rogerson, L.; et al. KDM4B is a Master Regulator of the Estrogen Receptor Signalling Cascade. Nucleic Acids Res. 2013, 41, 6892–6904. [Google Scholar] [CrossRef]
- Vleugel, M.; Shvarts, D.; Vanderwall, E.; Vandiest, P. P300 and P53 Levels Determine Activation of HIF-1 Downstream Targets in Invasive Breast Cancer. Hum. Pathol. 2006, 37, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, X.; Ding, T.; Zhang, H.; Zhang, Q.; Dai, H.; Zhang, H.; Tang, J.; Wang, X. KAT7 Promotes Radioresistance through Upregulating PI3K/AKT Signaling in Breast Cancer. J. Radiat. Res. 2023, 64, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.-J.; Dietel, M.; Weichert, W.; Denkert, C. Differential Expression of Histone Deacetylases HDAC1, 2 and 3 in Human Breast Cancer—Overexpression of HDAC2 and HDAC3 Is Associated with Clinicopathological Indicators of Disease Progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ding, S.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. HDAC1 Triggers the Proliferation and Migration of Breast Cancer Cells via Upregulation of Interleukin-8. Biol. Chem. 2017, 398, 1347–1356. [Google Scholar] [CrossRef]
- Shan, W.; Jiang, Y.; Yu, H.; Huang, Q.; Liu, L.; Guo, X.; Li, L.; Mi, Q.; Zhang, K.; Yang, Z. HDAC2 Overexpression Correlates with Aggressive Clinicopathological Features and DNA-Damage Response Pathway of Breast Cancer. Am. J. Cancer Res. 2017, 7, 1213–1226. [Google Scholar]
- Rahbari, R.; Rasmi, Y.; Khadem-Ansari, M.H.; Abdi, M. The Role of Histone Deacetylase 3 in Breast Cancer. Med. Oncol. 2022, 39, 84. [Google Scholar] [CrossRef]
- Kurani, H.; Razavipour, S.F.; Harikumar, K.B.; Dunworth, M.; Ewald, A.J.; Nasir, A.; Pearson, G.; Van Booven, D.; Zhou, Z.; Azzam, D.; et al. DOT1L Is a Novel Cancer Stem Cell Target for Triple-Negative Breast Cancer. Clin. Cancer Res. 2022, 28, 1948–1965. [Google Scholar] [CrossRef]
- Cho, M.-H.; Park, J.-H.; Choi, H.-J.; Park, M.-K.; Won, H.-Y.; Park, Y.-J.; Lee, C.H.; Oh, S.-H.; Song, Y.-S.; Kim, H.S.; et al. DOT1L Cooperates with the C-Myc-P300 Complex to Epigenetically Derepress CDH1 Transcription Factors in Breast Cancer Progression. Nat. Commun. 2015, 6, 7821. [Google Scholar] [CrossRef]
- Ghanbari, M.; Hosseinpour-Feizi, M.; Safaralizadeh, R.; Aghazadeh, A.; Montazeri, V. Study of KMT2B (MLL2) Gene Expression Changes in Patients with Breast Cancer. Breast Cancer Manag. 2019, 8, BMT24. [Google Scholar] [CrossRef]
- Gala, K.; Li, Q.; Sinha, A.; Razavi, P.; Dorso, M.; Sanchez-Vega, F.; Chung, Y.R.; Hendrickson, R.; Hsieh, J.J.; Berger, M.; et al. KMT2C Mediates the Estrogen Dependence of Breast Cancer through Regulation of ERα Enhancer Function. Oncogene 2018, 37, 4692–4710. [Google Scholar] [CrossRef]
- Jin, M.L.; Kim, Y.W.; Jin, H.L.; Kang, H.; Lee, E.K.; Stallcup, M.R.; Jeong, K.W. Aberrant Expression of SETD1A Promotes Survival and Migration of Estrogen Receptor A-positive Breast Cancer Cells. Int. J. Cancer 2018, 143, 2871–2883. [Google Scholar] [CrossRef]
- Jin, M.L.; Yang, L.; Jeong, K.W. SETD1A-SOX2 Axis Is Involved in Tamoxifen Resistance in Estrogen Receptor α-Positive Breast Cancer Cells. Theranostics 2022, 12, 5761–5775. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.-Y.; Park, M.K.; Choi, H.-J.; An, H.W.; Park, Y.-U.; Choi, H.-J.; Park, J.; Kim, H.-Y.; Son, T.; Lee, H.; et al. NSD3-Induced Methylation of H3K36 Activates NOTCH Signaling to Drive Breast Tumor Initiation and Metastatic Progression. Cancer Res. 2021, 81, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Park, S.; Park, S.-Y.; Lee, C.-Y.; Eum, D.-Y.; Shim, J.-W.; Choi, S.-H.; Choi, Y.-J.; Park, S.-J.; Heo, K. G9a Knockdown Suppresses Cancer Aggressiveness by Facilitating Smad Protein Phosphorylation through Increasing BMP5 Expression in Luminal A Type Breast Cancer. Int. J. Mol. Sci. 2022, 23, 589. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Sedukhina, A.S.; Nakagawa, Y.; Maeda, I.; Kubota, M.; Ohnuma, S.; Tsugawa, K.; Ohta, T.; Roche-Molina, M.; Bernal, J.A.; et al. LSD1 Overexpression Is Associated with Poor Prognosis in Basal-Like Breast Cancer, and Sensitivity to PARP Inhibition. PLoS ONE 2015, 10, e0118002. [Google Scholar] [CrossRef]
- Serce, N.; Gnatzy, A.; Steiner, S.; Lorenzen, H.; Kirfel, J.; Buettner, R. Elevated Expression of LSD1 (Lysine-Specific Demethylase 1) during Tumour Progression from Pre-Invasive to Invasive Ductal Carcinoma of the Breast. BMC Clin. Pathol. 2012, 12, 13. [Google Scholar] [CrossRef]
- Zheng, Q.; Fan, H.; Meng, Z.; Yuan, L.; Liu, C.; Peng, Y.; Zhao, W.; Wang, L.; Li, J.; Feng, J. Histone Demethylase KDM2B Promotes Triple Negative Breast Cancer Proliferation by Suppressing P15INK4B, P16INK4A, and P57KIP2 Transcription. Acta Biochim. Biophys. Sin. 2018, 50, 897–904. [Google Scholar] [CrossRef]
- Kottakis, F.; Foltopoulou, P.; Sanidas, I.; Keller, P.; Wronski, A.; Dake, B.T.; Ezell, S.A.; Shen, Z.; Naber, S.P.; Hinds, P.W.; et al. NDY1/KDM2B Functions as a Master Regulator of Polycomb Complexes and Controls Self-Renewal of Breast Cancer Stem Cells. Cancer Res. 2014, 74, 3935–3946. [Google Scholar] [CrossRef]
- Hou, J.; Wu, J.; Dombkowski, A.; Zhang, K.; Holowatyj, A.; Boerner, J.L.; Yang, Z.-Q. Genomic Amplification and a Role in Drug-Resistance for the KDM5A Histone Demethylase in Breast Cancer. Am. J. Transl. Res. 2012, 4, 247–256. [Google Scholar]
- Yamamoto, S.; Wu, Z.; Russnes, H.G.; Takagi, S.; Peluffo, G.; Vaske, C.; Zhao, X.; Moen Vollan, H.K.; Maruyama, R.; Ekram, M.B.; et al. JARID1B Is a Luminal Lineage-Driving Oncogene in Breast Cancer. Cancer Cell 2014, 25, 762–777. [Google Scholar] [CrossRef]
- Xun, J.; Gao, R.; Wang, B.; Li, Y.; Ma, Y.; Guan, J.; Zhang, Q. Histone Demethylase KDM6B Inhibits Breast Cancer Metastasis by Regulating Wnt/B-catenin Signaling. FEBS Open Bio 2021, 11, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Cai, M.; Chen, J.; Guan, X.; Kung, H.; Zeng, Y.; Xie, D. High Expression of P300 in Human Breast Cancer Correlates with Tumor Recurrence and Predicts Adverse Prognosis. Chin. J. Cancer Res. 2011, 23, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Fermento, M.E.; Gandini, N.A.; Salomón, D.G.; Ferronato, M.J.; Vitale, C.A.; Arévalo, J.; López Romero, A.; Nuñez, M.; Jung, M.; Facchinetti, M.M.; et al. Inhibition of P300 Suppresses Growth of Breast Cancer. Role of P300 Subcellular Localization. Exp. Mol. Pathol. 2014, 97, 411–424. [Google Scholar] [CrossRef]
- Ramadan, W.S.; Talaat, I.M.; Hachim, M.Y.; Lischka, A.; Gemoll, T.; El-Awady, R. The Impact of CBP Expression in Estrogen Receptor-Positive Breast Cancer. Clin. Epigenetics 2021, 13, 72. [Google Scholar] [CrossRef]
- Oh, J.H.; Lee, J.Y.; Kim, K.H.; Kim, C.Y.; Jeong, D.S.; Cho, Y.; Nam, K.T.; Kim, M.H. Elevated GCN5 Expression Confers Tamoxifen Resistance by Upregulating AIB1 Expression in ER-Positive Breast Cancer. Cancer Lett. 2020, 495, 145–155. [Google Scholar] [CrossRef]
- Judes, G.; Dubois, L.; Rifaï, K.; Idrissou, M.; Mishellany, F.; Pajon, A.; Besse, S.; Daures, M.; Degoul, F.; Bignon, Y.-J.; et al. TIP60: An Actor in Acetylation of H3K4 and Tumor Development in Breast Cancer. Epigenomics 2018, 10, 1415–1430. [Google Scholar] [CrossRef]
- Yu, L.; Liang, Y.; Cao, X.; Wang, X.; Gao, H.; Lin, S.-Y.; Schiff, R.; Wang, X.-S.; Li, K. Identification of MYST3 as a Novel Epigenetic Activator of ERα Frequently Amplified in Breast Cancer. Oncogene 2017, 36, 2910–2918. [Google Scholar] [CrossRef]
- Qiao, W.; Liu, H.; Liu, R.; Liu, Q.; Zhang, T.; Guo, W.; Li, P.; Deng, M. Prognostic and Clinical Significance of Histone Deacetylase 1 Expression in Breast Cancer: A Meta-Analysis. Clin. Chim. Acta 2018, 483, 209–215. [Google Scholar] [CrossRef]
- Cao, C.; Vasilatos, S.N.; Bhargava, R.; Fine, J.L.; Oesterreich, S.; Davidson, N.E.; Huang, Y. Functional Interaction of Histone Deacetylase 5 (HDAC5) and Lysine-Specific Demethylase 1 (LSD1) Promotes Breast Cancer Progression. Oncogene 2017, 36, 133–145. [Google Scholar] [CrossRef]
- Rahmani, G.; Sameri, S.; Abbasi, N.; Abdi, M.; Najafi, R. The Clinical Significance of Histone Deacetylase-8 in Human Breast Cancer. Pathol. Res. Pract. 2021, 220, 153396. [Google Scholar] [CrossRef]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 Promotes the Dissemination of Breast Cancer Cells via AKT/GSK-3β/Snail Signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef] [PubMed]
- Linares, A.; Assou, S.; Lapierre, M.; Thouennon, E.; Duraffourd, C.; Fromaget, C.; Boulahtouf, A.; Tian, G.; Ji, J.; Sahin, O.; et al. Increased Expression of the HDAC 9 Gene Is Associated with Antiestrogen Resistance of Breast Cancers. Mol. Oncol. 2019, 13, 1534–1547. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, X.-M.; Xiao, S.; Wu, Z.-R.; Shi, Y.-J.; Xie, M.-J. HDAC11 Is Related to Breast Cancer Prognosis and Inhibits Invasion and Proliferation of Breast Cancer Cells. Int. J. Clin. Exp. Pathol. 2023, 16, 172–183. [Google Scholar]
- Jin, X.; Wei, Y.; Xu, F.; Zhao, M.; Dai, K.; Shen, R.; Yang, S.; Zhang, N. SIRT1 Promotes Formation of Breast Cancer through Modulating Akt Activity. J. Cancer 2018, 9, 2012–2023. [Google Scholar] [CrossRef]
- Sarkar, S.; Venkatesh, D.; Kandasamy, T.; Ghosh, S.S. Epigenetic Modulations in Breast Cancer: An Emerging Paradigm in Therapeutic Implications. Front. Biosci. 2024, 29, 287. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic Regulation in Human Cancer: The Potential Role of Epi-Drug in Cancer Therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Ahmad, B.; Saeed, A.; Al-Amery, A.; Celik, I.; Ahmed, I.; Yaseen, M.; Khan, I.A.; Al-Fahad, D.; Bhat, M.A. Investigating Potential Cancer Therapeutics: Insight into Histone Deacetylases (HDACs) Inhibitions. Pharmaceuticals 2024, 17, 444. [Google Scholar] [CrossRef]
- Hu, Z.; Wei, F.; Su, Y.; Wang, Y.; Shen, Y.; Fang, Y.; Ding, J.; Chen, Y. Histone Deacetylase Inhibitors Promote Breast Cancer Metastasis by Elevating NEDD9 Expression. Signal Transduct. Target. Ther. 2023, 8, 11. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Bubna, A. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef]
- Palczewski, M.B.; Kuschman, H.P.; Bovee, R.; Hickok, J.R.; Thomas, D.D. Vorinostat Exhibits Anticancer Effects in Triple-Negative Breast Cancer Cells by Preventing Nitric Oxide-Driven Histone Deacetylation. Biol. Chem. 2021, 402, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Won, A.J.; Lee, J.; Jung, J.H.; Yoon, S.; Lee, B.M.; Kim, H.S. Molecular Mechanism of SAHA on Regulation of Autophagic Cell Death in Tamoxifen-Resistant MCF-7 Breast Cancer Cells. Int. J. Med. Sci. 2012, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting Triple-Negative Breast Cancer Cells with the Histone Deacetylase Inhibitor Panobinostat. Breast Cancer Res. 2012, 14, R79. [Google Scholar] [CrossRef]
- Matossian, M.D.; Burks, H.E.; Elliott, S.; Hoang, V.T.; Bowles, A.C.; Sabol, R.A.; Bunnell, B.A.; Martin, E.C.; Burow, M.E.; Collins-Burow, B.M. Panobinostat Suppresses the Mesenchymal Phenotype in a Novel Claudin-Low Triple Negative Patient-Derived Breast Cancer Model. Oncoscience 2018, 5, 99–108. [Google Scholar] [CrossRef]
- Fortunati, N.; Marano, F.; Bandino, A.; Frairia, R.; Catalano, M.G.; Boccuzzi, G. The Pan-Histone Deacetylase Inhibitor LBH589 (Panobinostat) Alters the Invasive Breast Cancer Cell Phenotype. Int. J. Oncol. 2014, 44, 700–708. [Google Scholar] [CrossRef]
- Lee, R.S.; Sad, K.; Fawwal, D.V.; Spangle, J.M. Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response. Cancers 2023, 15, 4005. [Google Scholar] [CrossRef]
- Yardley, D.A.; Ismail-Khan, R.; Klein, P. Results of ENCORE 301, a Randomized, Phase II, Double-Blind, Placebo-Controlled Study of Exemestane with or without Entinostat in Postmenopausal Women with Locally Recurrent or Metastatic Estrogen Receptor-Positive (ER+) Breast Cancer Progressing on a Non. J. Clin. Oncol. 2011, 29, 268. [Google Scholar] [CrossRef]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.-J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor–Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus Exemestane for Postmenopausal Patients with Advanced, Hormone Receptor-Positive Breast Cancer (ACE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.M.; Tan, P.B.O.; Liu, E.T.; Yu, Q. Pharmacologic Disruption of Polycomb-Repressive Complex 2-Mediated Gene Repression Selectively Induces Apoptosis in Cancer Cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Hayden, A.; Johnson, P.W.M.; Packham, G.; Crabb, S.J. S-Adenosylhomocysteine Hydrolase Inhibition by 3-Deazaneplanocin A Analogues Induces Anti-Cancer Effects in Breast Cancer Cell Lines and Synergy with Both Histone Deacetylase and HER2 Inhibition. Breast Cancer Res. Treat. 2011, 127, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep Is a Global Histone Methylation Inhibitor That Reactivates Developmental Genes Not Silenced by DNA Methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zheng, J.; Zou, J.X.; Xu, J.; Han, F.; Xiang, S.; Liu, P.; Chen, H.-W.; Wang, J. S-Adenosylhomocysteine (AdoHcy)-Dependent Methyltransferase Inhibitor DZNep Overcomes Breast Cancer Tamoxifen Resistance via Induction of NSD2 Degradation and Suppression of NSD2-Driven Redox Homeostasis. Chem. Biol. Interact. 2020, 317, 108965. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Gulati, N.; Béguelin, W.; Giulino-Roth, L. Enhancer of Zeste Homolog 2 (EZH2) Inhibitors. Leuk. Lymphoma 2018, 59, 1574–1585. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A.; Coindre, J.-M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 Inhibitor, in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma and Advanced Solid Tumours: A First-in-Human, Open-Label, Phase 1 Study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Nie, L.; Wei, Y.; Zhang, F.; Hsu, Y.-H.; Chan, L.-C.; Xia, W.; Ke, B.; Zhu, C.; Deng, R.; Tang, J.; et al. CDK2-Mediated Site-Specific Phosphorylation of EZH2 Drives and Maintains Triple-Negative Breast Cancer. Nat. Commun. 2019, 10, 5114. [Google Scholar] [CrossRef]
- Brown, L.J.; Achinger-Kawecka, J.; Portman, N.; Clark, S.; Stirzaker, C.; Lim, E. Epigenetic Therapies and Biomarkers in Breast Cancer. Cancers 2022, 14, 474. [Google Scholar] [CrossRef]
- Chi, K.R. The Dark Side of the Human Genome. Nature 2016, 538, 275–277. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin Signature Reveals over a Thousand Highly Conserved Large Non-Coding RNAs in Mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Bonadio, R.S.; Buson, L.; Chemello, F.; Cagnin, S. A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 302. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Cech, T.R. The Recruitment of Chromatin Modifiers by Long Noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Mandal, S.S. LncRNA HOTAIR: A Master Regulator of Chromatin Dynamics and Cancer. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 151–164. [Google Scholar] [CrossRef]
- Rani, V.; Sengar, R.S. Biogenesis and Mechanisms of MicroRNA-mediated Gene Regulation. Biotechnol. Bioeng. 2022, 119, 685–692. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, Y.; Zhai, Y.; Ma, X.; An, X.; Zhang, S.; Li, Z. MicroRNA-29b Regulates DNA Methylation by Targeting Dnmt3a/3b and Tet1/2/3 in Porcine Early Embryo Development. Dev. Growth Differ. 2018, 60, 197–204. [Google Scholar] [CrossRef]
- Xie, H.; Liu, Y.; Du, R.; Wang, B.; Chen, M.; Zhang, Y.; Deng, Z.; Li, J. MiR-377 Induces Senescence in Human Skin Fibroblasts by Targeting DNA Methyltransferase 1. Cell Death Dis. 2017, 8, e2663. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, S.; Li, N.; Yang, X.; Ma, S.; Yang, A.; Zhang, H.; Yang, S.; Mao, C.; Xu, L.; et al. MiR-34a Targets HDAC1-Regulated H3K9 Acetylation on Lipid Accumulation Induced by Homocysteine in Foam Cells. J. Cell. Biochem. 2017, 118, 4617–4627. [Google Scholar] [CrossRef]
- OuYang, C.; Shu, G.; Liu, J.; Deng, S.; Lu, P.; Li, Y.; Gan, Y.; Xie, B.; Liu, J.; Yin, G. HDAC5, Negatively Regulated by miR-148a-3p, Promotes Colon Cancer Cell Migration. Cancer Sci. 2022, 113, 2560–2574. [Google Scholar] [CrossRef]
- Zhang, P.-P.; Wang, X.; Zhao, W.; Qi, B.; Yang, Q.; Wan, H.-Y.; Shuang, Z.; Liu, M.; Li, X.; Li, S.; et al. DNA Methylation-Mediated Repression of MiR-941 Enhances Lysine (K)-Specific Demethylase 6B Expression in Hepatoma Cells. J. Biol. Chem. 2014, 289, 24724–24735. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Ye, P.; Murai, K.; Lang, M.-F.; Li, S.; Zhang, H.; Li, W.; Fu, C.; Yin, J.; Wang, A.; et al. MiR-137 Forms a Regulatory Loop with Nuclear Receptor TLX and LSD1 in Neural Stem Cells. Nat. Commun. 2011, 2, 529. [Google Scholar] [CrossRef] [PubMed]
- Guajardo, L.; Aguilar, R.; Bustos, F.J.; Nardocci, G.; Gutiérrez, R.A.; van Zundert, B.; Montecino, M. Downregulation of the Polycomb-Associated Methyltransferase Ezh2 during Maturation of Hippocampal Neurons Is Mediated by MicroRNAs Let-7 and MiR-124. Int. J. Mol. Sci. 2020, 21, 8472. [Google Scholar] [CrossRef] [PubMed]
- Dal-Pra, S.; Hodgkinson, C.P.; Mirotsou, M.; Kirste, I.; Dzau, V.J. Demethylation of H3K27 Is Essential for the Induction of Direct Cardiac Reprogramming by MiR Combo. Circ. Res. 2017, 120, 1403–1413. [Google Scholar] [CrossRef]
- Han, F.; Huang, D.; Meng, J.; Chu, J.; Wang, M.; Chen, S. MiR-126-5p Enhances Radiosensitivity of Lung Adenocarcinoma Cells by Inhibiting EZH2 via the KLF2/BIRC Axis. J. Cell. Mol. Med. 2022, 26, 2529–2542. [Google Scholar] [CrossRef]
- Wade, S.L.; Langer, L.F.; Ward, J.M.; Archer, T.K. MiRNA-Mediated Regulation of the SWI/SNF Chromatin Remodeling Complex Controls Pluripotency and Endodermal Differentiation in Human ESCs. Stem Cells 2015, 33, 2925–2935. [Google Scholar] [CrossRef]
- Lusser, A.; Kadonaga, J.T. Chromatin Remodeling by ATP-dependent Molecular Machines. BioEssays 2003, 25, 1192–1200. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Esteller, M. Non-Coding RNAs, Epigenetics, and Cancer: Tying It All Together. Cancer Metastasis Rev. 2018, 37, 55–73. [Google Scholar] [CrossRef]
- Amicone, L.; Marchetti, A.; Cicchini, C. The LncRNA HOTAIR: A Pleiotropic Regulator of Epithelial Cell Plasticity. J. Exp. Clin. Cancer Res. 2023, 42, 147. [Google Scholar] [CrossRef]
- Xin, X.; Li, Q.; Fang, J.; Zhao, T. LncRNA HOTAIR: A Potential Prognostic Factor and Therapeutic Target in Human Cancers. Front. Oncol. 2021, 11, 679244. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Li, P.; Wang, J.; Shu, Y.; Zhong, X.; Gao, Z.; Yang, J.; Jiang, Y.; Zhou, X.; et al. Long Noncoding RNA HOTAIR Regulates the Stemness of Breast Cancer Cells via Activation of the NF-KB Signaling Pathway. J. Biol. Chem. 2022, 298, 102630. [Google Scholar] [CrossRef]
- Cantile, M.; Di Bonito, M.; Cerrone, M.; Collina, F.; De Laurentiis, M.; Botti, G. Long Non-Coding RNA HOTAIR in Breast Cancer Therapy. Cancers 2020, 12, 1197. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long Non-Coding RNA HOTAIR Reprograms Chromatin State to Promote Cancer Metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The Degradation of EZH2 Mediated by LncRNA ANCR Attenuated the Invasion and Metastasis of Breast Cancer. Cell Death Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef]
- Pan, Y.; Li, C.; Chen, J.; Zhang, K.; Chu, X.; Wang, R.; Chen, L. The Emerging Roles of Long Noncoding RNA ROR (LincRNA-ROR) and Its Possible Mechanisms in Human Cancers. Cell. Physiol. Biochem. 2016, 40, 219–229. [Google Scholar] [CrossRef]
- Hu, A.; Hong, F.; Li, D.; Jin, Y.; Kon, L.; Xu, Z.; He, H.; Xie, Q. Long Non-Coding RNA ROR Recruits Histone Transmethylase MLL1 to up-Regulate TIMP3 Expression and Promote Breast Cancer Progression. J. Transl. Med. 2021, 19, 95. [Google Scholar] [CrossRef]
- Kim, J.; Siverly, A.N.; Chen, D.; Wang, M.; Yuan, Y.; Wang, Y.; Lee, H.; Zhang, J.; Muller, W.J.; Liang, H.; et al. Ablation of MiR-10b Suppresses Oncogene-Induced Mammary Tumorigenesis and Metastasis and Reactivates Tumor-Suppressive Pathways. Cancer Res. 2016, 76, 6424–6435. [Google Scholar] [CrossRef]
- Hossain, A.; Kuo, M.T.; Saunders, G.F. Mir-17-5p Regulates Breast Cancer Cell Proliferation by Inhibiting Translation of AIB1 MRNA. Mol. Cell. Biol. 2006, 26, 8191–8201. [Google Scholar] [CrossRef]
- Mohammaddoust, S.; Sadeghizadeh, M. Mir-183 Functions as an Oncogene via Decreasing PTEN in Breast Cancer Cells. Sci. Rep. 2023, 13, 8086. [Google Scholar] [CrossRef]
- Wu, X. Expressions of MiR-21 and MiR-210 in Breast Cancer and Their Predictive Values for Prognosis. Iran. J. Public Health 2020, 49, 21. [Google Scholar] [CrossRef]
- Rao, X.; Di Leva, G.; Li, M.; Fang, F.; Devlin, C.; Hartman-Frey, C.; Burow, M.E.; Ivan, M.; Croce, C.M.; Nephew, K.P. MicroRNA-221/222 Confers Breast Cancer Fulvestrant Resistance by Regulating Multiple Signaling Pathways. Oncogene 2011, 30, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.M.; Zeinelabdeen, Y.; Manie, T.; Khallaf, E.; Assal, R.A.; Youness, R.A. MiR-17–5p/STAT3/H19: A Novel Regulatory Axis Tuning ULBP2 Expression in Young Breast Cancer Patients. Pathol. Res. Pract. 2024, 263, 155638. [Google Scholar] [CrossRef]
- Pan, Z.; Niu, G.; Cao, C.; Tian, Y. Role of MicroRNAs in Remodeling the Tumor Microenvironment (Review). Int. J. Oncol. 2020, 56, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, Y.; Xu, S.; Jiang, Y.; Yuan, C.; Zhou, L.; Ma, X.; Bai, Y.; Lu, J.; Ma, J. DEPDC1, Negatively Regulated by miR-26b, Facilitates Cell Proliferation via the up-Regulation of FOXM1 Expression in TNBC. Cancer Lett. 2019, 442, 242–251. [Google Scholar] [CrossRef]
- Yan, G.; Li, Y.; Zhan, L.; Sun, S.; Yuan, J.; Wang, T.; Yin, Y.; Dai, Z.; Zhu, Y.; Jiang, Z.; et al. Decreased MiR-124-3p Promoted Breast Cancer Proliferation and Metastasis by Targeting MGAT5. Am. J. Cancer Res. 2019, 9, 585–596. [Google Scholar]
- Sibilano, M.; Tullio, V.; Adorno, G.; Savini, I.; Gasperi, V.; Catani, M.V. Platelet-Derived MiR-126-3p Directly Targets AKT2 and Exerts Anti-Tumor Effects in Breast Cancer Cells: Further Insights in Platelet-Cancer Interplay. Int. J. Mol. Sci. 2022, 23, 5484. [Google Scholar] [CrossRef]
- Gambacurta, A.; Tullio, V.; Savini, I.; Mauriello, A.; Catani, M.V.; Gasperi, V. Identification of the EBF1/ETS2/KLF2-miR-126-Gene Feed-Forward Loop in Breast Carcinogenesis and Stemness. Int. J. Mol. Sci. 2025, 26, 328. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, X.; Shen, C.; Shi, Y. MicroRNA-203 Suppresses Cell Proliferation and Migration by Targeting BIRC5 and LASP1 in Human Triple-Negative Breast Cancer Cells. J. Exp. Clin. Cancer Res. 2012, 31, 58. [Google Scholar] [CrossRef]
- Wang, L.; Kang, F.; Wang, J.; Yang, C.; He, D. Downregulation of MiR-205 Contributes to Epithelial–Mesenchymal Transition and Invasion in Triple-Negative Breast Cancer by Targeting HMGB1–RAGE Signaling Pathway. Anticancer Drugs 2019, 30, 225–232. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Y.; Fan, X.; Zhang, P.; Wang, P.; Cheng, S.; Zhang, J. MicroRNA-125b as a Tumor Suppressor by Targeting MMP11 in Breast Cancer. Thorac. Cancer 2020, 11, 1613–1620. [Google Scholar] [CrossRef]
- Nie, J.; Jiang, H.C.; Zhou, Y.C.; Jiang, B.; He, W.J.; Wang, Y.F.; Dong, J. MiR-125b Regulates the Proliferation and Metastasis of Triple Negative Breast Cancer Cells via the Wnt/β-Catenin Pathway and EMT. Biosci. Biotechnol. Biochem. 2019, 83, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.P.; Pérez-Moreno, P.; Pérez, Y.; Calaf, G.M. The Role of MicroRNAs in Breast Cancer and the Challenges of Their Clinical Application. Diagnostics 2023, 13, 3072. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Z. The Emerging Role of MicroRNAs in Breast Cancer. J. Oncol. 2020, 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, Y.; Zhou, X. The Roles of MicroRNAs in Epigenetic Regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Humphries, B.; Wang, Z.; Yang, C. MicroRNA Regulation of Epigenetic Modifiers in Breast Cancer. Cancers 2019, 11, 897. [Google Scholar] [CrossRef]
- Denis, H.; Van Grembergen, O.; Delatte, B.; Dedeurwaerder, S.; Putmans, P.; Calonne, E.; Rothé, F.; Sotiriou, C.; Fuks, F.; Deplus, R. MicroRNAs Regulate KDM5 Histone Demethylases in Breast Cancer Cells. Mol. Biosyst. 2016, 12, 404–413. [Google Scholar] [CrossRef]
- Li, H.; Li, H.-H.; Chen, Q.; Wang, Y.-Y.; Fan, C.-C.; Duan, Y.-Y.; Huang, Y.; Zhang, H.-M.; Li, J.-P.; Zhang, X.-Y.; et al. miR-142-5p Inhibits Cell Invasion and Migration by Targeting DNMT1 in Breast Cancer. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2021, 28, 885–897. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Chen, Y.-B.; Yue, H.-R.; Zhou, X.-J.; Ma, H.-Y.; Wang, X.; Cao, X.-C.; Yu, Y. PAX5-MiR-142 Feedback Loop Promotes Breast Cancer Proliferation by Regulating DNMT1 and ZEB1. Mol. Med. 2023, 29, 89. [Google Scholar] [CrossRef]
- Ng, E.K.O.; Li, R.; Shin, V.Y.; Siu, J.M.; Ma, E.S.K.; Kwong, A. MicroRNA-143 Is Downregulated in Breast Cancer and Regulates DNA Methyltransferases 3A in Breast Cancer Cells. Tumor Biol. 2014, 35, 2591–2598. [Google Scholar] [CrossRef]
- Wu, M.; Fan, B.; Guo, Q.; Li, Y.; Chen, R.; Lv, N.; Diao, Y.; Luo, Y. Knockdown of SETDB1 Inhibits Breast Cancer Progression by MiR-381-3p-Related Regulation. Biol. Res. 2018, 51, 39. [Google Scholar] [CrossRef]
- Tang, H.; Liu, P.; Yang, L.; Xie, X.; Ye, F.; Wu, M.; Liu, X.; Chen, B.; Zhang, L.; Xie, X. MiR-185 Suppresses Tumor Proliferation by Directly Targeting E2F6 and DNMT1 and Indirectly Upregulating BRCA1 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Lei, Y.; Liu, X. MiR-29a Promotes Cell Proliferation and EMT in Breast Cancer by Targeting Ten Eleven Translocation 1. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Wang, D.; Wei, X. MiR-22 Suppresses Tumorigenesis and Improves Radiosensitivity of Breast Cancer Cells by Targeting Sirt1. Biol. Res. 2017, 50, 27. [Google Scholar] [CrossRef]
- Wang, P.; Li, Z.; Liu, H.; Zhou, D.; Fu, A.; Zhang, E. MicroRNA-126 Increases Chemosensitivity in Drug-Resistant Gastric Cancer Cells by Targeting EZH2. Biochem. Biophys. Res. Commun. 2016, 479, 91–96. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Liang, Y.; Zhao, M.; Long, H.; Ding, S.; Yin, H.; Lu, Q. MicroRNA-126 Regulates DNA Methylation in CD4+ T Cells and Contributes to Systemic Lupus Erythematosus by Targeting DNA Methyltransferase 1. Arthritis Rheum. 2011, 63, 1376–1386. [Google Scholar] [CrossRef]
- Wang, K.; Yang, F.; Men, X.; Li, G.; Sun, C. MiR-138 Suppresses EMT through Degradation KDM6B in Breast Carcinoma. Int. J. Clin. Exp. Med. 2016, 9, 4724–4733. [Google Scholar]
- Liu, B.; Zhang, X.; Song, F.; Zheng, H.; Zhao, Y.; Li, H.; Zhang, L.; Yang, M.; Zhang, W.; Chen, K. MiR-502/SET8 Regulatory Circuit in Pathobiology of Breast Cancer. Cancer Lett. 2016, 376, 259–267. [Google Scholar] [CrossRef]
- Zeng, Z.; Yang, Y.; Wu, H. MicroRNA-765 Alleviates the Malignant Progression of Breast Cancer via Interacting with EZH1. Am. J. Transl. Res. 2019, 11, 4500–4507. [Google Scholar]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-Antagonism Regulates Breast Cancer Stemness and Metastasis via TET-Family-Dependent Chromatin Remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef]
- Mekala, J.R.; Naushad, S.M.; Ponnusamy, L.; Arivazhagan, G.; Sakthiprasad, V.; Pal-Bhadra, M. Epigenetic Regulation of miR-200 as the Potential Strategy for the Therapy Against Triple-Negative Breast Cancer. Gene 2018, 641, 248–258. [Google Scholar] [CrossRef]
- Pang, Y.; Liu, J.; Li, X.; Xiao, G.; Wang, H.; Yang, G.; Li, Y.; Tang, S.; Qin, S.; Du, N.; et al. MYC and DNMT 3A-mediated DNA Methylation Represses Micro RNA-200b in Triple Negative Breast Cancer. J. Cell. Mol. Med. 2018, 22, 6262–6274. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Li, Y.; Qian, X.; Hu, Y.; Liu, J.; Zhang, S.; Zhang, J. MiR-340 Inhibits Triple-Negative Breast Cancer Progression by Reversing EZH2 Mediated MiRNAs Dysregulated Expressions. J. Cancer 2017, 8, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Noyan, S.; Andac Ozketen, A.; Gurdal, H.; Gur Dedeoglu, B. MiR-770-5p Regulates EMT and Invasion in TNBC Cells by Targeting DNMT3A. Cell. Signal. 2021, 83, 109996. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, W.; Tang, T.; Wang, Y.; Yin, X.; Chen, Y.; Zhang, Y.; Xing, Y.; Shen, Y.; Xia, T.; et al. MiR-29a Contributes to Breast Cancer Cells Epithelial–Mesenchymal Transition, Migration, and Invasion via down-Regulating Histone H4K20 Trimethylation through Directly Targeting SUV420H2. Cell Death Dis. 2019, 10, 176. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, Y.; Yang, M.; Jat, P.; Li, K.; Lombardo, Y.; Xiong, D.; Coombes, R.C.; Raguz, S.; Yagüe, E. The MiR-106b∼25 Cluster Promotes Bypass of Doxorubicin-Induced Senescence and Increase in Motility and Invasion by Targeting the E-Cadherin Transcriptional Activator EP300. Cell Death Differ. 2014, 21, 462–474. [Google Scholar] [CrossRef]
- Ao, X.; Nie, P.; Wu, B.; Xu, W.; Zhang, T.; Wang, S.; Chang, H.; Zou, Z. Decreased Expression of MicroRNA-17 and MicroRNA-20b Promotes Breast Cancer Resistance to Taxol Therapy by Upregulation of NCOA3. Cell Death Dis. 2016, 7, e2463. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Fu, J.; Xiao, X.; Wu, J.; Wu, R.-C. MiR-34a Regulates Therapy Resistance by Targeting HDAC1 and HDAC7 in Breast Cancer. Cancer Lett. 2014, 354, 311–319. [Google Scholar] [CrossRef]
- Mocavini, I.; Pippa, S.; Licursi, V.; Paci, P.; Trisciuoglio, D.; Mannironi, C.; Presutti, C.; Negri, R. JARID1B Expression and its Function in DNA Damage Repair Are Tightly Regulated by MiRNAs in Breast Cancer. Cancer Sci. 2019, 110, 1232–1243. [Google Scholar] [CrossRef]
- Ahmad, A.; Ginnebaugh, K.R.; Yin, S.; Bollig-Fischer, A.; Reddy, K.B.; Sarkar, F.H. Functional Role of MiR-10b in Tamoxifen Resistance of ER-Positive Breast Cancer Cells through down-Regulation of HDAC4. BMC Cancer 2015, 15, 540. [Google Scholar] [CrossRef]
- Xu, Q.; Jiang, Y.; Yin, Y.; Li, Q.; He, J.; Jing, Y.; Qi, Y.-T.; Xu, Q.; Li, W.; Lu, B.; et al. A Regulatory Circuit of MiR-148a/152 and DNMT1 in Modulating Cell Transformation and Tumor Angiogenesis through IGF-IR and IRS1. J. Mol. Cell Biol. 2013, 5, 3–13. [Google Scholar] [CrossRef]
- Gurbuz, V.; Sozen, S.; Bilen, C.; Konac, E. MiR-148a, MiR-152 and MiR-200b Promote Prostate Cancer Metastasis by Targeting DNMT1 and PTEN Expression. Oncol. Lett. 2021, 22, 805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, X.-X.; He, J.-R.; Zhou, C.-X.; Guo, M.; He, M.; Li, M.-F.; Chen, G.-Q.; Zhao, Q. Pathologically Decreased MiR-26a Antagonizes Apoptosis and Facilitates Carcinogenesis by Targeting MTDH and EZH2 in Breast Cancer. Carcinogenesis 2011, 32, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.; Hsu, C.-Y.; Tsai, C.-F.; Long, C.-Y.; Wu, C.-H.; Wu, D.-C.; Lee, J.-N.; Chang, W.-C.; Tsai, E.-M. HDAC Inhibitors Target HDAC5, Upregulate MicroRNA-125a-5p, and Induce Apoptosis in Breast Cancer Cells. Mol. Ther. 2015, 23, 656–666. [Google Scholar] [CrossRef]
- Abdolvahabi, Z.; Nourbakhsh, M.; Hosseinkhani, S.; Hesari, Z.; Alipour, M.; Jafarzadeh, M.; Ghorbanhosseini, S.S.; Seiri, P.; Yousefi, Z.; Yarahmadi, S.; et al. MicroRNA-590-3P Suppresses Cell Survival and Triggers Breast Cancer Cell Apoptosis via Targeting Sirtuin-1 and Deacetylation of P53. J. Cell. Biochem. 2019, 120, 9356–9368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, Inhibited Indirectly by LincRNA HOTAIR, Directly Inhibits SETDB1 and Reverses the EMT of Breast Cancer Stem Cells by Downregulating the STAT3 Pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef]
- Ma, W.; Xiao, G.G.; Mao, J.; Lu, Y.; Song, B.; Wang, L.; Fan, S.; Fan, P.; Hou, Z.; Li, J.; et al. Dysregulation of the MiR-34a-SIRT1 Axis Inhibits Breast Cancer Stemness. Oncotarget 2015, 6, 10432–10444. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of MiR-200 Inhibition of Suz12 Leads to Polycomb-Mediated Repression Required for the Formation and Maintenance of Cancer Stem Cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef]
- Roscigno, G.; Quintavalle, C.; Donnarumma, E.; Puoti, I.; Diaz-Lagares, A.; Iaboni, M.; Fiore, D.; Russo, V.; Todaro, M.; Romano, G.; et al. MiR-221 Promotes Stemness of Breast Cancer Cells by Targeting DNMT3b. Oncotarget 2016, 7, 580–592. [Google Scholar] [CrossRef]
- Hui, Z.; Yiling, C.; Wenting, Y.; XuQun, H.; ChuanYi, Z.; Hui, L. MiR-491-5p Functions as a Tumor Suppressor by Targeting JMJD2B in ERα-positive Breast Cancer. FEBS Lett. 2015, 589, 812–821. [Google Scholar] [CrossRef]
- Perri, P.; Ponzoni, M.; Corrias, M.V.; Ceccherini, I.; Candiani, S.; Bachetti, T. A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma. Cancers 2021, 13, 5528. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, C.; Liu, X.; Wu, C.; Yin, H. Long Non-Coding RNA HOTAIR Enhances Radioresistance in MDA-MB231 Breast Cancer Cells. Oncol. Lett. 2017, 13, 1143–1148. [Google Scholar] [CrossRef]
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 Locus: Role of an Imprinted Non-coding RNA in Growth and Development. BioEssays 2010, 32, 473–480. [Google Scholar] [CrossRef]
- Hashemi, M.; Moosavi, M.S.; Abed, H.M.; Dehghani, M.; Aalipour, M.; Heydari, E.A.; Behroozaghdam, M.; Entezari, M.; Salimimoghadam, S.; Gunduz, E.S.; et al. Long Non-Coding RNA (LncRNA) H19 in Human Cancer: From Proliferation and Metastasis to Therapy. Pharmacol. Res. 2022, 184, 106418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Weaver, D.L.; Olsen, D.; DeKay, J.; Peng, Z.; Ashikaga, T.; Evans, M.F. Long Non-Coding RNA Chromogenic In Situ Hybridisation Signal Pattern Correlation with Breast Tumour Pathology. J. Clin. Pathol. 2016, 69, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xie, S.; Yang, J.; Xiong, H.; Jia, Y.; Zhou, Y.; Chen, Y.; Ying, X.; Chen, C.; Ye, C.; et al. The Long Noncoding RNA H19 Promotes Tamoxifen Resistance in Breast Cancer via Autophagy. J. Hematol. Oncol. 2019, 12, 81. [Google Scholar] [CrossRef]
- Garcia-Padilla, C.; Lozano-Velasco, E.; Muñoz-Gallardo, M.D.M.; Castillo-Casas, J.M.; Caño-Carrillo, S.; Martínez-Amaro, F.J.; García-López, V.; Aránega, A.; Franco, D.; García-Martínez, V.; et al. LncRNA H19 Impairs Chemo and Radiotherapy in Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 8309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 LncRNA Alters DNA Methylation Genome Wide by Regulating S-Adenosylhomocysteine Hydrolase. Nat. Commun. 2015, 6, 10221. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Salvatore, M.; Incoronato, M. MiRNA-Based Therapeutics in Breast Cancer: A Systematic Review. Front. Oncol. 2021, 11, 668464. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Cho, S.K. Targeting MiRNAs by Histone Deacetylase Inhibitors (HDACi): Rationalizing Epigenetics-Based Therapies for Breast Cancer. Pharmacol. Ther. 2020, 206, 107437. [Google Scholar] [CrossRef]
- Li, L.; Xie, X.; Luo, J.; Liu, M.; Xi, S.; Guo, J.; Kong, Y.; Wu, M.; Gao, J.; Xie, Z.; et al. Targeted Expression of MiR-34a Using the T-VISA System Suppresses Breast Cancer Cell Growth and Invasion. Mol. Ther. 2012, 20, 2326–2334. [Google Scholar] [CrossRef]
- Adams, B.D.; Wali, V.B.; Cheng, C.J.; Inukai, S.; Booth, C.J.; Agarwal, S.; Rimm, D.L.; Győrffy, B.; Santarpia, L.; Pusztai, L.; et al. MiR-34a Silences c-SRC to Attenuate Tumor Growth in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic Silencing of MiR-10b Inhibits Metastasis in a Mouse Mammary Tumor Model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liang, Z.; Feng, A.; Salgado, E.; Shim, H. HDAC Inhibitor Suppresses Proliferation and Invasion of Breast Cancer Cells through Regulation of MiR-200c Targeting CRKL. Biochem. Pharmacol. 2018, 147, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Trapani, D.; Esposito, A.; Criscitiello, C.; Mazzarella, L.; Locatelli, M.; Minchella, I.; Minucci, S.; Curigliano, G. Entinostat for the Treatment of Breast Cancer. Expert Opin. Investig. Drugs 2017, 26, 965–971. [Google Scholar] [CrossRef]
- Schech, A.; Kazi, A.; Yu, S.; Shah, P.; Sabnis, G. Histone Deacetylase Inhibitor Entinostat Inhibits Tumor-Initiating Cells in Triple-Negative Breast Cancer Cells. Mol. Cancer Ther. 2015, 14, 1848–1857. [Google Scholar] [CrossRef]
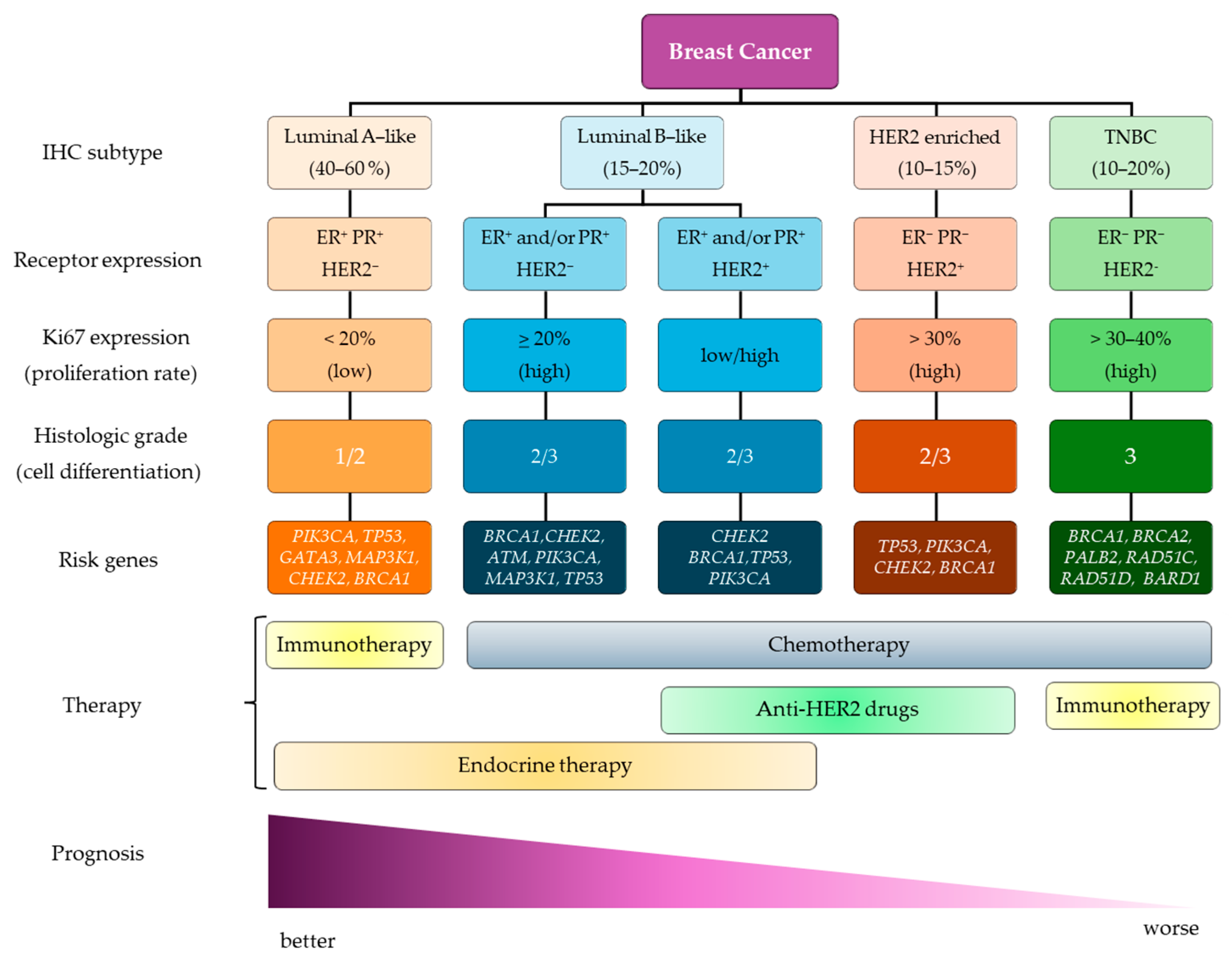

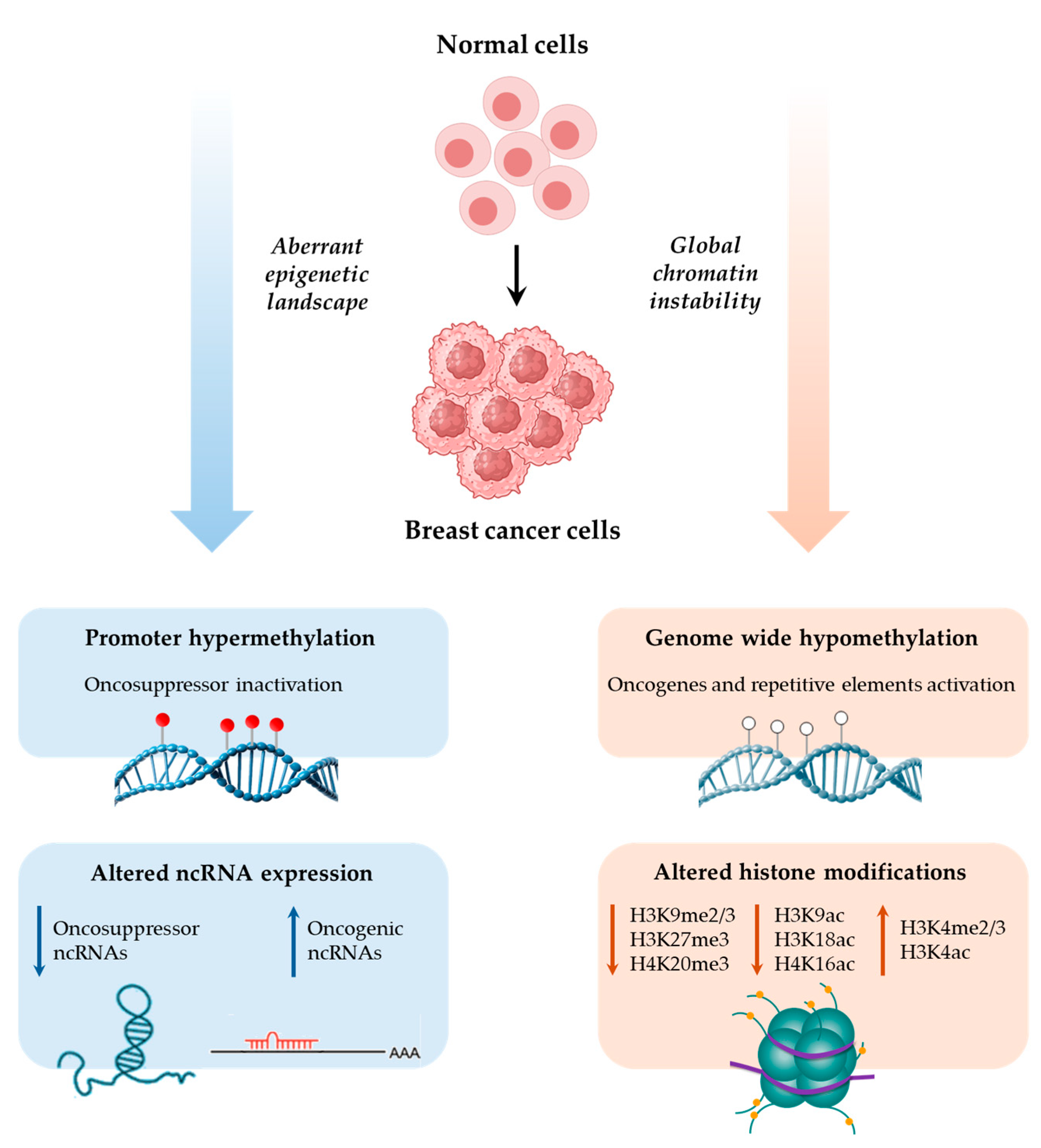
| Gene | Main Findings | Clinical Relevance | Ref. |
|---|---|---|---|
| Hypermethylated genes | |||
| BRCA1 | Positively related to tumor grade, ER and PR status | Predictive | [45] |
| No correlation with clinicopathological features | - | [46] | |
| Associated with better prognosis and chemotherapy efficacy | Prognostic | [47] | |
| Related to grade, Ki67, and HER2 levels | Diagnostic/prognostic | [48] | |
| MGMT | Related to lymph node involvement, grade and stage, and ER and PR loss | Therapy response | [49] |
| Associated with TNBC | Therapy response | [50] | |
| E-cadherin | Positively correlated with ER expression, lymph node metastasis, and poor overall and disease-free survival | Prognostic | [51,52] |
| RARβ2 | Hypermethylation marks the transition from proliferative epithelial hyperplasia towards atypia, occurring especially during transition from DH to ADH | Risk biomarker | [53,54,55] |
| Cyclin D2 | Related to poor prognosis | Diagnostic/prognostic | [56] |
| RASSF1A | Related to tumor size, ER and PR status, immunohistochemical subtype | Prognostic Therapy response | [57] |
| Hypermethylation marks the transition from normal epithelia towards DH | Risk biomarker | [54] | |
| CDKN2A (p14ARF/p16INK4a) | p14ARF methylation related to peritumoral vessel involvement, p53 mutations and PR− | Prognostic | [58] |
| p16INK4a methylation negatively associated with ER, PR, and HER2 expression | Diagnostic | [59] | |
| ERα | Related to resistance to endocrine therapy | Therapy response | [60,61] |
| PTEN | Related to tumor size, histologic grade, ER and HER2 status, and disease-free and overall survival | Prognostic | [62,63,64] |
| APC | Hypermethylation marks the transition from healthy tissue to benign lesions to BC; no correlation with clinicopathological features | Risk biomarker | [55] |
| Related to chemotherapy, distant metastasis, and overall survival | Prognostic | [65] | |
| Hypomethylated genes | |||
| LINE-1 | Associated with tumor stage, lymph node metastasis, older age, distant recurrence, and disease-free and overall survival | Prognostic | [66] |
| CXCR4 | Correlated with tumor stage and size, histological grade, lymph node status, metastasis, and death | Prognostic | [67] |
| miRNAs (target) | Biological Role | Dysregulation | Refs | |
|---|---|---|---|---|
| Proliferation | miR-17-5p (KAT13B) | Inhibition of proliferation | ↓ in BC | [230] |
| miR-137 (KDM5B) | Inhibition of proliferation and migration | [248] | ||
| miR-138 (KDM5C) | ||||
| miR-142-5p (DNMT1) | [249,250] | |||
| miR-143 (DNMT3A) | [251] | |||
| miR-381-3p (SETDB1) | [252] | |||
| miR-185 (DNMT1) | Inhibition of proliferation by indirectly up-regulating BRCA1 expression | ↓ in TNBC | [253] | |
| miR-29a (TET1) | Increase in proliferation, migration, and EMT; negatively correlated with poor survival | ↑ in ER− BC | [254] | |
| EMT, invasion, migration, metastasis | miR-22 (SIRT1) | Inhibition of tumorigenesis and improvement of radiosensitivity | ↓ in BC | [255] |
| miR-126 (EZH2, DNMT1) | Inhibition of proliferation, EMT, invasion, and metastatic potential | [239,256,257] | ||
| miR-138 (KDM6B) | Inhibition of EMT and invasion; associated with lymph node metastasis, TNM stage, and poor prognosis | [258] | ||
| miR-502 (KMT5A) | Inhibition of proliferation, invasion, migration, and EMT | [259] | ||
| miR-765 (EZH1) | Inhibition of proliferative, migratory and invasive abilities; related to tumor stage, metastasis, metastasis, and poor prognosis | [260] | ||
| miR-22 (TET1/2/3) | It indirectly targets TET family members by antagonizing miR-200b; related to stemness, EMT, invasion, metastasis, and poor clinical outcomes | [261] | ||
| miR-200b (DNMT3A) | Inhibition of EMT | ↓ in TNBC | [262,263] | |
| miR-340 (EZH2) | Inhibition of cell growth, invasion and migration, and induction of apoptosis | [264] | ||
| miR-770-5p (DNMT3A) | Inhibition of EMT and invasion | [265] | ||
| miR-29 (SUV420H2) | Stimulation of EMT, migration, and invasion | ↑ in BC stem cells | [266] | |
| miR-29a (TET1) | Stimulation of EMT, proliferation, and migration | ↑ in ER− BC | [254] | |
| miR-25, miR-93, miR-106b (P300) | Stimulation of invasion, migration, and EMT | ↑ in BC | [267] | |
| Therapy resistance | miR-17, miR-20b (KAT13B) | Stimulation of chemosensitivity | ↓ in taxol-resistant cells | [268] |
| miR-22 (SIRT1) | Inhibition of tumorigenesis and stimulation of radiosensitivity | ↓ in BC | [254] | |
| miR-34a (HDAC1, HDAC7) | Stimulation of therapy sensitivity; negatively correlated with tumor grade and stage | [269] | ||
| miR-486 (KDM5B) | Involved in DNA damage repair and radiosensitivity | [270] | ||
| miR-10b (HDAC4) | Associated with invasiveness, migration, and tamoxifen-resistance | ↑ in tamoxifen-resistant cells | [271] | |
| Angiogenesis | miR-148a, miR-152 (DNMT1) | Inhibition of tumor growth and angiogenesis by down-regulating IGF-IR and IRS1 | ↓ in BC | [272,273] |
| Apoptosis | miR-26a (EZH2) | Inhibition of proliferation and induction of apoptosis | [274] | |
| miR-125a-5p (HDAC5) | Stimulation of apoptosis in BC stem cells by targeting apoptosis-related genes | [242,275] | ||
| miR-590-3p (SIRT1) | Stimulation of apoptosis | [276] | ||
| Stemness | miR-7 (SETDB1) | Reverses the EMT of BC stem cells by downregulating the STAT3 pathway | ↓ in BC stem cells | [277] |
| miR-34a (SIRT1) | Inhibition of stemness markers | [278] | ||
| miR-200b (SUZ12) | Regulation of E-cadherin expression and stemness | [279] | ||
| miR-221, miR-222 (DNMT3B) | Stimulation of expression of pluripotency-associated genes (Nanog, Oct 3/4) | ↑ in BC stem cells | [280] | |
| Estrogen signaling | miR-491-5p (KDM4B) | Inhibition of estrogen signaling and estrogen-stimulated proliferation | ↓ in ERα+ BC | [281] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortellesi, E.; Savini, I.; Veneziano, M.; Gambacurta, A.; Catani, M.V.; Gasperi, V. Decoding the Epigenome of Breast Cancer. Int. J. Mol. Sci. 2025, 26, 2605. https://doi.org/10.3390/ijms26062605
Cortellesi E, Savini I, Veneziano M, Gambacurta A, Catani MV, Gasperi V. Decoding the Epigenome of Breast Cancer. International Journal of Molecular Sciences. 2025; 26(6):2605. https://doi.org/10.3390/ijms26062605
Chicago/Turabian StyleCortellesi, Elisa, Isabella Savini, Matteo Veneziano, Alessandra Gambacurta, Maria Valeria Catani, and Valeria Gasperi. 2025. "Decoding the Epigenome of Breast Cancer" International Journal of Molecular Sciences 26, no. 6: 2605. https://doi.org/10.3390/ijms26062605
APA StyleCortellesi, E., Savini, I., Veneziano, M., Gambacurta, A., Catani, M. V., & Gasperi, V. (2025). Decoding the Epigenome of Breast Cancer. International Journal of Molecular Sciences, 26(6), 2605. https://doi.org/10.3390/ijms26062605