
Heart Failure and Arrhythmias: Circadian and Epigenetic Interplay in Myocardial Electrophysiology

Abstract
1. Introduction
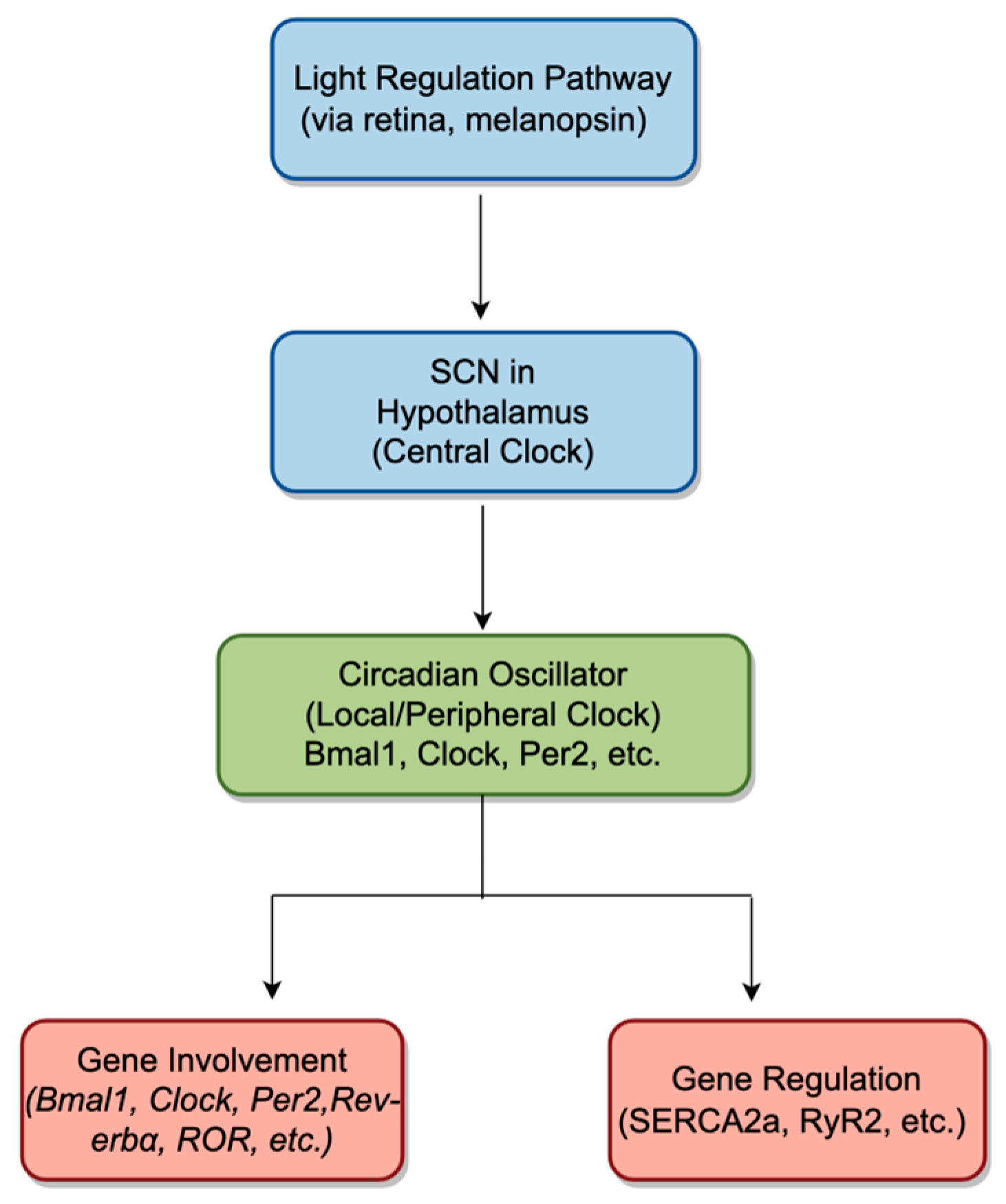
2. The Circadian Clock’s Roles in Cardiac Electrophysiology
2.1. Circadian Rhythms in Myocardial Cells
2.1.1. Overview of Circadian Rhythms
2.1.2. Role of Circadian Rhythms in the Heart
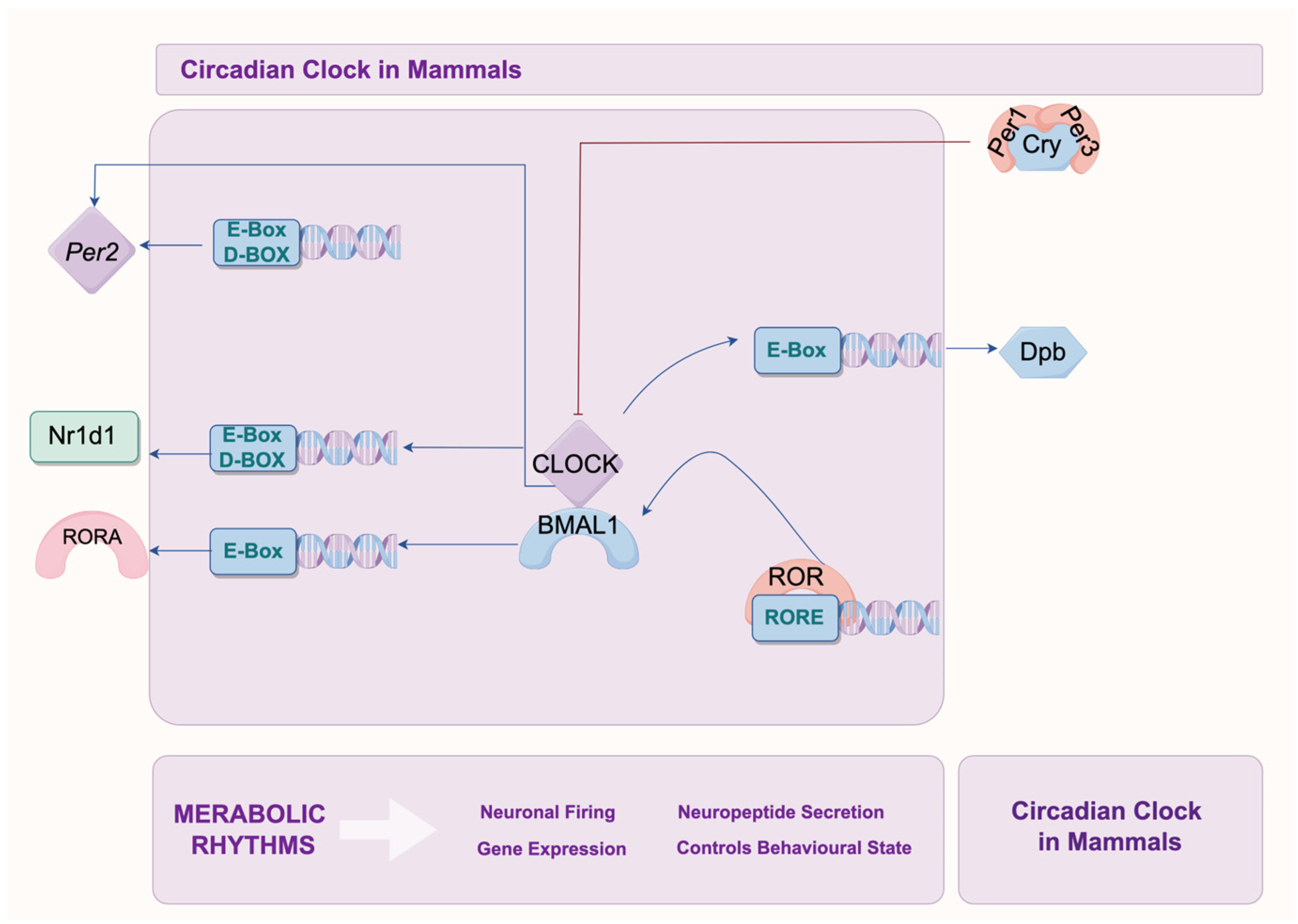
2.2. Core Clock Genes and Their Mechanisms
2.3. Circadian Regulation of Ion Channels and Transporters
2.3.1. Circadian Rhythm and Myocardial Depolarization
2.3.2. Circadian Rhythm and Myocardial Repolarization
2.3.3. Impact on Electromechanical Activity in Myocardial Cell
3. Effects of Circadian Clock Proteins in Modulating Myocardial Electrophysiology
3.1. Circadian Regulation of Ion Channels
3.1.1. Sodium Channel
3.1.2. Potassium Channels
3.1.3. Calcium Channels
4. Clinical Implications and Therapeutic Potential Approaches
4.1. Overview of Current Treatments for Heart Failure-Related Arrhythmias
4.1.1. Pharmacological Therapy
4.1.2. Device-Based Therapy
4.1.3. Procedural Interventions
4.1.4. Lifestyle Modifications
4.1.5. Education and Monitoring
4.2. Potential Interventions Targeting Circadian Rhythm Proteins and Epigenetic Pathways
4.3. Chrono-Pharmacology Represents a Promising Therapeutic Approach
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baman, J.R.; Ahmad, F.S. Heart Failure. JAMA 2020, 324, 1015. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef]
- Zou, S.; Wang, Z.; Bhura, M.; Zhang, G.; Tang, K. Prevalence and associated socioeconomic factors of multimorbidity in 10 regions of China: An analysis of 0.5 million adults. J. Public Health 2022, 44, 36–50. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar]
- Kumar, A.; Avishay, D.M.; Jones, C.R.; Shaikh, J.D.; Kaur, R.; Aljadah, M.; Kichloo, A.; Shiwalkar, N.; Keshavamurthy, S. Sudden cardiac death: Epidemiology, pathogenesis and management. Rev. Cardiovasc. Med. 2021, 22, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Sudden cardiac death in China: Current status and future perspectives. Europace 2015, 17 (Suppl. S2), ii14–ii18. [Google Scholar] [CrossRef]
- Stiles, M.K.; Wilde, A.A.M.; Abrams, D.J.; Ackerman, M.J.; Albert, C.M.; Behr, E.R.; Chugh, S.S.; Cornel, M.C.; Gardner, K.; Ingles, J.; et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm. 2021, 18, e1–e50. [Google Scholar] [CrossRef]
- Hsia, H.H.; Xiong, N. Mapping and Ablation of Ventricular Arrhythmias in Cardiomyopathies. Card. Electrophysiol. Clin. 2019, 11, 635–655. [Google Scholar] [CrossRef]
- Gaztañaga, L.; Marchlinski, F.E.; Betensky, B.P. Mechanisms of cardiac arrhythmias. Rev. Esp. Cardiol. 2012, 65, 174–185. [Google Scholar] [CrossRef]
- Meregalli, P.G.; Wilde, A.A.; Tan, H.L. Pathophysiological mechanisms of Brugada syndrome: Depolarization disorder, repolarization disorder, or more? Cardiovasc. Res. 2005, 67, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.C.; Wu, W.T.; Chen, Y.C.; Yang, F.M.; Tsai, W.C.; Lee, C.H. Betel-Quid Chewing, Heart Failure, and Premature Ventricular Contractions in Patients with Cardiopulmonary Symptoms. Int. J. Environ. Res. Public Health 2020, 17, 7472. [Google Scholar] [CrossRef]
- AlMahameed, S.T.; Ziv, O. Ventricular Arrhythmias. Med. Clin. N. Am. 2019, 103, 881–895. [Google Scholar] [CrossRef]
- Sozzi, F.B.; Gherbesi, E.; Faggiano, A.; Gnan, E.; Maruccio, A.; Schiavone, M.; Iacuzio, L.; Carugo, S. Viral Myocarditis: Classification, Diagnosis, and Clinical Implications. Front. Cardiovasc. Med. 2022, 9, 908663. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Smith, S.A.; Makara, M.A.; Mohler, P.J. Chapter 7—Cellular and Molecular Pathobiology of the Cardiac Conduction System. In Cellular and Molecular Pathobiology of Cardiovascular Disease; Willis, M.S., Homeister, J.W., Stone, J.R., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 121–134. [Google Scholar]
- Moore, J.P.; Khairy, P. Adults with Congenital Heart Disease and Arrhythmia Management. Cardiol. Clin. 2020, 38, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Morton, J.B.; Davidson, N.C.; Spence, S.J.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Electrical remodeling of the atria in congestive heart failure: Electrophysiological and electroanatomic mapping in humans. Circulation 2003, 108, 1461–1468. [Google Scholar] [CrossRef]
- Gilman, N.V. Analysis for science librarians of the 2017 Nobel prize in physiology or medicine: The life and work of Jeffrey C. Hall, Michael Rosbash, and Michael W. Young. Sci. Technol. Libr. 2018, 37, 22–47. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Z.; Shi, G.; Xing, L.; Wang, X.; Gu, X.; Qu, Z.; Dong, Z.; Xiong, J.; Gao, X.; et al. The circadian clock influences heart performance. J. Biol. Rhythm. 2011, 26, 402–411. [Google Scholar] [CrossRef]
- Crnko, S.; Du Pre, B.C.; Sluijter, J.P.G.; Van Laake, L.W. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat. Rev. Cardiol. 2019, 16, 437–447. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, H.; Huang, S.; Yin, L.; Wang, F.; Luo, P.; Huang, H. Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2022, 7, 200. [Google Scholar] [CrossRef]
- Salvador, R.B.; Tomotani, B.M. Clocks at a snail pace: Biological rhythms in terrestrial gastropods. PeerJ 2024, 12, e18318. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.M.; Loros, J.J.; Dunlap, J.C. Circadian Oscillators: Around the Transcription-Translation Feedback Loop and on to Output. Trends Biochem. Sci. 2016, 41, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Robinson, I.; Reddy, A.B. Molecular mechanisms of the circadian clockwork in mammals. FEBS Lett. 2014, 588, 2477–2483. [Google Scholar] [CrossRef]
- Walker, W.H., 2nd; Walton, J.C.; DeVries, A.C.; Nelson, R.J. Circadian rhythm disruption and mental health. Transl. Psychiatry 2020, 10, 28. [Google Scholar] [CrossRef]
- Natale, V.; Andreose, A.; Bacaro, V.; Giovagnoli, S.; Giudetti, F.; Grimaldi, M.; Tonetti, L.; Crocetti, E. Morningness–Eveningness Preference and Motor Wake–Sleep Inertia in Adolescents. Sensors 2024, 24, 7668. [Google Scholar] [CrossRef]
- Hou, T.; Guo, Z.; Gong, M.C. Circadian variations of vasoconstriction and blood pressure in physiology and diabetes. Curr. Opin. Pharmacol. 2021, 57, 125–131. [Google Scholar] [CrossRef]
- Chen, L.; Yang, G. Recent advances in circadian rhythms in cardiovascular system. Front. Pharmacol. 2015, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Black, N.; D’Souza, A.; Wang, Y.; Piggins, H.; Dobrzynski, H.; Morris, G.; Boyett, M.R. Circadian rhythm of cardiac electrophysiology, arrhythmogenesis, and the underlying mechanisms. Heart Rhythm. 2019, 16, 298–307. [Google Scholar] [CrossRef]
- Koronowski, K.B.; Sassone-Corsi, P. Communicating clocks shape circadian homeostasis. Science 2021, 371, eabd0951. [Google Scholar] [CrossRef]
- Lu, Q.; Kim, J.Y. Mammalian circadian networks mediated by the suprachiasmatic nucleus. FEBS J. 2022, 289, 6589–6604. [Google Scholar] [CrossRef]
- Scheer, F.A.; Ter Horst, G.J.; van Der Vliet, J.; Buijs, R.M. Physiological and anatomic evidence for regulation of the heart by suprachiasmatic nucleus in rats. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1391–H1399. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, J.; Fahrenkrug, J. Melanopsin containing retinal ganglion cells are light responsive from birth. Neuroreport 2004, 15, 2317–2320. [Google Scholar] [CrossRef] [PubMed]
- Le Minh, N.; Damiola, F.; Tronche, F.; Schutz, G.; Schibler, U. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J. 2001, 20, 7128–7136. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Nakamura, T.J.; Nakamura, W.; Tokuda, I.T. Modeling circadian regulation of ovulation timing: Age-related disruption of estrous cyclicity. Sci. Rep. 2020, 10, 16767. [Google Scholar] [CrossRef]
- Cailotto, C.; Lei, J.; van der Vliet, J.; van Heijningen, C.; van Eden, C.G.; Kalsbeek, A.; Pevet, P.; Buijs, R.M. Effects of nocturnal light on (clock) gene expression in peripheral organs: A role for the autonomic innervation of the liver. PLoS ONE 2009, 4, e5650. [Google Scholar] [CrossRef]
- D’Souza, A.; Wang, Y.; Anderson, C.; Bucchi, A.; Baruscotti, M.; Olieslagers, S.; Mesirca, P.; Johnsen, A.B.; Mastitskaya, S.; Ni, H.; et al. A circadian clock in the sinus node mediates day-night rhythms in Hcn4 and heart rate. Heart Rhythm. 2021, 18, 801–810. [Google Scholar] [CrossRef]
- Cavey, M.; Collins, B.; Bertet, C.; Blau, J. Circadian rhythms in neuronal activity propagate through output circuits. Nat. Neurosci. 2016, 19, 587–595. [Google Scholar] [CrossRef]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 2011, 4, 362–372. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, L.; Sun, L.; Li, Y.; Gao, Y.; Xu, C.; Shao, Y.; Li, M.; Li, C.; Lu, Y.; et al. LncRNA ZFAS1 as a SERCA2a Inhibitor to Cause Intracellular Ca(2+) Overload and Contractile Dysfunction in a Mouse Model of Myocardial Infarction. Circ. Res. 2018, 122, 1354–1368. [Google Scholar] [CrossRef]
- Muslimova, E.; Rebrova, T.Y.; Kondratieva, D.; Akhmedov, S.D.; Afanasiev, S. Expression of the Ca2+-ATPase SERCA2a (ATP2A2) gene and the ryanodine receptor (RYR2) gene in patients with chronic heart failure. Russ. J. Genet. 2020, 56, 843–848. [Google Scholar] [CrossRef]
- Lee, C.; Etchegaray, J.P.; Cagampang, F.R.; Loudon, A.S.; Reppert, S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Toh, K.L.; Jones, C.R.; He, Y.; Eide, E.J.; Hinz, W.A.; Virshup, D.M.; Ptacek, L.J.; Fu, Y.H. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 2001, 291, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Padiath, Q.S.; Shapiro, R.E.; Jones, C.R.; Wu, S.C.; Saigoh, N.; Saigoh, K.; Ptacek, L.J.; Fu, Y.H. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 2005, 434, 640–644. [Google Scholar] [CrossRef]
- Preitner, N.; Damiola, F.; Lopez-Molina, L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Singh, K.; Jha, N.K.; Thakur, A. Spatiotemporal chromatin dynamics—A telltale of circadian epigenetic gene regulation. Life Sci. 2019, 221, 377–391. [Google Scholar] [CrossRef]
- Battaglin, F.; Chan, P.; Pan, Y.; Soni, S.; Qu, M.; Spiller, E.R.; Castanon, S.; Roussos Torres, E.T.; Mumenthaler, S.M.; Kay, S.A.; et al. Clocking cancer: The circadian clock as a target in cancer therapy. Oncogene 2021, 40, 3187–3200. [Google Scholar] [CrossRef]
- Kojetin, D.J.; Burris, T.P. REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov. 2014, 13, 197–216. [Google Scholar] [CrossRef]
- Gavillet, B.; Rougier, J.S.; Domenighetti, A.A.; Behar, R.; Boixel, C.; Ruchat, P.; Lehr, H.A.; Pedrazzini, T.; Abriel, H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 2006, 99, 407–414. [Google Scholar] [CrossRef]
- Jeyaraj, D.; Haldar, S.M.; Wan, X.; McCauley, M.D.; Ripperger, J.A.; Hu, K.; Lu, Y.; Eapen, B.L.; Sharma, N.; Ficker, E.; et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 2012, 483, 96–99. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, D.; Yuan, J.; Han, Z.; Wang, Y.; Qian, Z.; Hou, X.; Wu, T.; Zou, J. CLOCK-BMAL1 regulate the cardiac L-type calcium channel subunit CACNA1C through PI3K-Akt signaling pathway. Can. J. Physiol. Pharmacol. 2016, 94, 1023–1032. [Google Scholar] [CrossRef]
- Schroder, E.A.; Burgess, D.E.; Manning, C.L.; Zhao, Y.; Moss, A.J.; Patwardhan, A.; Elayi, C.S.; Esser, K.A.; Delisle, B.P. Light phase-restricted feeding slows basal heart rate to exaggerate the type-3 long QT syndrome phenotype in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1777–H1785. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Sekiguchi, A.; Iwasaki, Y.K.; Sagara, K.; Iinuma, H.; Hatano, S.; Fu, L.T.; Watanabe, H. Circadian variation of cardiac K+ channel gene expression. Circulation 2003, 107, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Plante, A.E.; Whitt, J.P.; Meredith, A.L. BK channel activation by L-type Ca(2+) channels Ca(V)1.2 and Ca(V)1.3 during the subthreshold phase of an action potential. J. Neurophysiol. 2021, 126, 427–439. [Google Scholar] [CrossRef]
- Marchal, G.A.; Remme, C.A. Subcellular diversity of Nav1.5 in cardiomyocytes: Distinct functions, mechanisms and targets. J. Physiol. 2023, 601, 941–960. [Google Scholar] [CrossRef] [PubMed]
- Pierre, M.; Djemai, M.; Poulin, H.; Chahine, M. Na(V)1.5 knockout in iPSCs: A novel approach to study Na(V)1.5 variants in a human cardiomyocyte environment. Sci. Rep. 2021, 11, 17168. [Google Scholar] [CrossRef]
- Song, W.; Shou, W. Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr. Cardiol. 2012, 33, 943–949. [Google Scholar] [CrossRef]
- Schroder, E.A.; Lefta, M.; Zhang, X.; Bartos, D.C.; Feng, H.Z.; Zhao, Y.; Patwardhan, A.; Jin, J.P.; Esser, K.A.; Delisle, B.P. The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility. Am. J. Physiol. Cell Physiol. 2013, 304, C954–C965. [Google Scholar] [CrossRef]
- Peters, S.; Thompson, B.A.; Perrin, M.; James, P.; Zentner, D.; Kalman, J.M.; Vandenberg, J.I.; Fatkin, D. Arrhythmic Phenotypes Are a Defining Feature of Dilated Cardiomyopathy-Associated SCN5A Variants: A Systematic Review. Circ. Genom. Precis. Med. 2022, 15, e003432. [Google Scholar] [CrossRef]
- Amoni, M.; Dries, E.; Ingelaere, S.; Vermoortele, D.; Roderick, H.L.; Claus, P.; Willems, R.; Sipido, K.R. Ventricular Arrhythmias in Ischemic Cardiomyopathy-New Avenues for Mechanism-Guided Treatment. Cells 2021, 10, 2629. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, W.; Wang, L.; Rane, M.J.; Han, F.; Cai, L. Multiple roles of KLF15 in the heart: Underlying mechanisms and therapeutic implications. J. Mol. Cell Cardiol. 2019, 129, 193–196. [Google Scholar] [CrossRef]
- Zhang, L.; Prosdocimo, D.A.; Bai, X.; Fu, C.; Zhang, R.; Campbell, F.; Liao, X.; Coller, J.; Jain, M.K. KLF15 Establishes the Landscape of Diurnal Expression in the Heart. Cell Rep. 2015, 13, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Schroder, E.A.; Burgess, D.E.; Zhang, X.; Lefta, M.; Smith, J.L.; Patwardhan, A.; Bartos, D.C.; Elayi, C.S.; Esser, K.A.; Delisle, B.P. The cardiomyocyte molecular clock regulates the circadian expression of Kcnh2 and contributes to ventricular repolarization. Heart Rhythm. 2015, 12, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Schroder, E.A.; Ono, M.; Johnson, S.R.; Rozmus, E.R.; Burgess, D.E.; Esser, K.A.; Delisle, B.P. The role of the cardiomyocyte circadian clocks in ion channel regulation and cardiac electrophysiology. J. Physiol. 2022, 600, 2037–2048. [Google Scholar] [CrossRef]
- Joukar, S. A comparative review on heart ion channels, action potentials and electrocardiogram in rodents and human: Extrapolation of experimental insights to clinic. Lab. Anim. Res. 2021, 37, 25. [Google Scholar] [CrossRef] [PubMed]
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: Ionic and non-ionic mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570. [Google Scholar] [CrossRef]
- Schroder, E.A.; Wayland, J.L.; Samuels, K.M.; Shah, S.F.; Burgess, D.E.; Seward, T.; Elayi, C.S.; Esser, K.A.; Delisle, B.P. Cardiomyocyte Deletion of Bmal1 Exacerbates QT- and RR-Interval Prolongation in Scn5a (+/ΔKPQ) Mice. Front. Physiol. 2021, 12, 681011. [Google Scholar] [CrossRef]
- Miceli, F.; Soldovieri, M.V.; Weckhuysen, S.; Cooper, E.; Taglialatela, M. KCNQ2-Related Disorders. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhang, Y.; Yao, C.; Ju, Z.; Jiao, D.; Hu, D.; Qi, L.; Liu, S.; Wu, X.; Zhao, C. Krüppel-like factors in tumors: Key regulators and therapeutic avenues. Front. Oncol. 2023, 13, 1080720. [Google Scholar] [CrossRef]
- Xie, Z.; Su, W.; Liu, S.; Zhao, G.; Esser, K.; Schroder, E.A.; Lefta, M.; Stauss, H.M.; Guo, Z.; Gong, M.C. Smooth-muscle BMAL1 participates in blood pressure circadian rhythm regulation. J. Clin. Investig. 2015, 125, 324–336. [Google Scholar] [CrossRef]
- Ávalos Prado, P.; Häfner, S.; Comoglio, Y.; Wdziekonski, B.; Duranton, C.; Attali, B.; Barhanin, J.; Sandoz, G. KCNE1 is an auxiliary subunit of two distinct ion channel superfamilies. Cell 2021, 184, 534–544.e11. [Google Scholar] [CrossRef]
- Hayter, E.A.; Wehrens, S.M.T.; Van Dongen, H.P.A.; Stangherlin, A.; Gaddameedhi, S.; Crooks, E.; Barron, N.J.; Venetucci, L.A.; O’Neill, J.S.; Brown, T.M.; et al. Distinct circadian mechanisms govern cardiac rhythms and susceptibility to arrhythmia. Nat. Commun. 2021, 12, 2472. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, J.; Qin, Y.; Wang, J.; Zhou, L. Mutations in voltage-gated L-type calcium channel: Implications in cardiac arrhythmia. Channels 2018, 12, 201–218. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Han, L.; Wang, Y.; Zhou, Y.; Li, Q.; Wu, Y.; Talabieke, S.; Hou, Y.; Wu, L.; et al. Functional Calsequestrin-1 Is Expressed in the Heart and Its Deficiency Is Causally Related to Malignant Hyperthermia-Like Arrhythmia. Circulation 2021, 144, 788–804. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- Naccarelli, G.V.; Lukas, M.A. Carvedilol’s antiarrhythmic properties: Therapeutic implications in patients with left ventricular dysfunction. Clin. Cardiol. 2005, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Diamond, A.; Goldenberg, I.; Younis, A.; Goldenberg, I.; Sampath, R.; Kutyifa, V.; Chen, A.Y.; McNitt, S.; Polonsky, B.; Steinberg, J.S.; et al. Effect of Carvedilol vs Metoprolol on Atrial and Ventricular Arrhythmias Among Implantable Cardioverter-Defibrillator Recipients. JACC Clin. Electrophysiol. 2023, 9, 2122–2131. [Google Scholar] [CrossRef]
- Kotecha, D.; Bunting, K.V.; Gill, S.K.; Mehta, S.; Stanbury, M.; Jones, J.C.; Haynes, S.; Calvert, M.J.; Deeks, J.J.; Steeds, R.P.; et al. Effect of Digoxin vs Bisoprolol for Heart Rate Control in Atrial Fibrillation on Patient-Reported Quality of Life: The RATE-AF Randomized Clinical Trial. JAMA 2020, 324, 2497–2508. [Google Scholar] [CrossRef]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, L.; Mohanty, P.; Mohanty, S.; Santangeli, P.; Trivedi, C.; Lakkireddy, D.; Reddy, M.; Jais, P.; Themistoclakis, S.; Dello Russo, A.; et al. Ablation Versus Amiodarone for Treatment of Persistent Atrial Fibrillation in Patients With Congestive Heart Failure and an Implanted Device: Results From the AATAC Multicenter Randomized Trial. Circulation 2016, 133, 1637–1644. [Google Scholar] [CrossRef]
- Torp-Pedersen, C.; Møller, M.; Bloch-Thomsen, P.E.; Køber, L.; Sandøe, E.; Egstrup, K.; Agner, E.; Carlsen, J.; Videbaek, J.; Marchant, B.; et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N. Engl. J. Med. 1999, 341, 857–865. [Google Scholar] [CrossRef]
- Rousseau, M.F.; Massart, P.E.; van Eyll, C.; Etienne, J.; Ahn, S.; Ghadanfar, M.; Friedrich, T.; Pouleur, H. Cardiac and hemodynamic effects of intravenous dofetilide in patients with heart failure. Am. J. Cardiol. 2001, 87, 1250–1254. [Google Scholar] [CrossRef]
- Gottlieb, S.S.; Singh, S.; Munger, M.; Eichhorn, E.J.; Ilgenfritz, J.; Hanyok, J. Hemodynamic effects of the class III antiarrhythmic drug, d-sotalol, in patients with congestive heart failure. Am. J. Cardiol. 1996, 78, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.; Ozierański, K.; Balsam, P.; Dąbrowski, R.; Farkowski, M.M.; Gackowski, A.; Jędrzejczyk-Patej, E.; Kalarus, Z.; Leszek, P.; Nessler, J.; et al. The effect of sacubitril/valsartan on the occurrence of ventricular arrhythmia and the risk of sudden cardiac death in patients with chronic heart failure with reduced left ventricular ejection fraction. Expert opinion of the Heart Rhythm and Heart Failure Sections of the Polish Cardiac Society. Pol. Heart J. 2019, 77, 987–993. [Google Scholar]
- Cohen, J.B.; Schrauben, S.J.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Yarde, M.; Wang, Z.; Bhattacharya, P.T.; Chirinos, D.A.; et al. Clinical Phenogroups in Heart Failure With Preserved Ejection Fraction: Detailed Phenotypes, Prognosis, and Response to Spironolactone. JACC Heart Fail. 2020, 8, 172–184. [Google Scholar] [CrossRef]
- Naser, N.; Durak-Nalbantic, A.; Sabanovic-Bajramovic, N.; Karic, A. The Effectiveness of Eplerenone vs Spironolactone on Left Ventricular Systolic Function, Hospitalization and Cardiovascular Death in Patients With Chronic Heart Failure-HFrEF. Med. Arch. 2023, 77, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Mentz, R.J.; Anstrom, K.J.; Eisenstein, E.L.; Sapp, S.; Greene, S.J.; Morgan, S.; Testani, J.M.; Harrington, A.H.; Sachdev, V.; Ketema, F.; et al. Effect of Torsemide vs Furosemide After Discharge on All-Cause Mortality in Patients Hospitalized With Heart Failure: The TRANSFORM-HF Randomized Clinical Trial. JAMA 2023, 329, 214–223. [Google Scholar] [CrossRef]
- Wojcik, C.; Warden, B.A. Mechanisms and Evidence for Heart Failure Benefits from SGLT2 Inhibitors. Curr. Cardiol. Rep. 2019, 21, 130. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, e263–e421. [Google Scholar]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients With Heart Failure: Proposal of a Novel Mechanism of Action. JAMA Cardiol. 2017, 2, 1025–1029. [Google Scholar] [CrossRef]
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef]
- Packer, M.; Butler, J.; Filippatos, G.S.; Jamal, W.; Salsali, A.; Schnee, J.; Kimura, K.; Zeller, C.; George, J.; Brueckmann, M.; et al. Evaluation of the effect of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality of patients with chronic heart failure and a reduced ejection fraction: Rationale for and design of the EMPEROR-Reduced trial. Eur. J. Heart Fail. 2019, 21, 1270–1278. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Borlaug, B.A.; Kitzman, D.W.; Shah, S.J.; Tang, F.; Khariton, Y.; Malik, A.O.; Khumri, T.; Umpierrez, G.; et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: A multicenter randomized trial. Nat. Med. 2021, 27, 1954–1960. [Google Scholar] [CrossRef]
- Khand, A.U.; Rankin, A.C.; Martin, W.; Taylor, J.; Gemmell, I.; Cleland, J.G. Carvedilol alone or in combination with digoxin for the management of atrial fibrillation in patients with heart failure? J. Am. Coll. Cardiol. 2003, 42, 1944–1951. [Google Scholar] [CrossRef]
- Mariani, M.V.; Lavalle, C.; Palombi, M.; Pierucci, N.; Trivigno, S.; D’Amato, A.; Filomena, D.; Cipollone, P.; Laviola, D.; Piro, A.; et al. SGLT2i reduce arrhythmic events in heart failure patients with cardiac implantable electronic devices. ESC Heart Fail. 2025. [Google Scholar] [CrossRef] [PubMed]
- Kuck, K.H.; Cappato, R.; Siebels, J.; Rüppel, R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation 2000, 102, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Paccione, E.N.; Lange, M.; Orkild, B.A.; Bergquist, J.A.; Kwan, E.; Hunt, B.; Dosdall, D.; Macleod, R.S.; Ranjan, R. Effects of Biventricular Pacing Locations on Anti-Tachycardia Pacing Success in a Patient-Specific Model. In Proceedings of the 2023 Computing in Cardiology, Atlanta, GA, USA, 1–4 October 2023. [Google Scholar]
- Friedman, P.; Murgatroyd, F.; Boersma, L.V.A.; Manlucu, J.; O’Donnell, D.; Knight, B.P.; Clémenty, N.; Leclercq, C.; Amin, A.; Merkely, B.P.; et al. Efficacy and Safety of an Extravascular Implantable Cardioverter-Defibrillator. N. Engl. J. Med. 2022, 387, 1292–1302. [Google Scholar] [CrossRef]
- Tankut, S.; Goldenberg, I.; Kutyifa, V.; Zareba, W.; Bragazzi, N.L.; McNitt, S.; Huang, D.T.; Aktas, M.K.; Younis, A. Cardiac resynchronization therapy and ventricular tachyarrhythmia burden. Heart Rhythm. 2021, 18, 762–769. [Google Scholar] [CrossRef]
- Deogharia, M.; Gurha, P. Epigenetic regulation of heart failure. Curr. Opin. Cardiol. 2024, 39, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.L.; Cervantes, M.; Froufe, H.J.C.; Egas, C.; Cunha-Oliveira, T.; Sassone-Corsi, P.; Oliveira, P.J. Doxorubicin persistently rewires cardiac circadian homeostasis in mice. Arch. Toxicol. 2020, 94, 257–271. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Xu, L.W.; Ding, S.; Ji, Q.Q.; Lin, N.; He, Q.; Gao, L.C.; Su, Y.Y.; Pu, J.; He, B. Nuclear receptor retinoid-related orphan receptor α deficiency exacerbates high-fat diet-induced cardiac dysfunction despite improving metabolic abnormality. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1991–2000. [Google Scholar] [CrossRef]
- Berulava, T.; Buchholz, E.; Elerdashvili, V.; Pena, T.; Islam, M.R.; Lbik, D.; Mohamed, B.A.; Renner, A.; von Lewinski, D.; Sacherer, M.; et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020, 22, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Koczor, C.A.; Ludlow, I.; Hight, R.S., 2nd; Jiao, Z.; Fields, E.; Ludaway, T.; Russ, R.; Torres, R.A.; Lewis, W. Ecstasy (MDMA) Alters Cardiac Gene Expression and DNA Methylation: Implications for Circadian Rhythm Dysfunction in the Heart. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 148, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Saleem, U.; Iman, S.; Akhtar, M.F.; Saleem, A.; Anwar, F.; Ahmad, B. Chronopharmacology: Appraising the Influence of Biorhythms on the Efficacy and Safety of Antihypertensive Drugs. Crit. Rev. Eukaryot. Gene Exp. 2019, 29, 499–509. [Google Scholar] [CrossRef]
- Nagata, N.; Kawasumi, M.; Fujimura, A.; Ando, H. Edoxaban Dosing Time Affects Blood Coagulation Inhibition in Rats. TH Open 2021, 5, e107–e112. [Google Scholar] [CrossRef]
- Diekman, C.O.; Wei, N. Circadian Rhythms of Early Afterdepolarizations and Ventricular Arrhythmias in a Cardiomyocyte Model. Biophys. J. 2021, 120, 319–333. [Google Scholar] [CrossRef]
- Kaskal, M.; Sevim, M.; Ulker, G.; Keles, C.; Bebitoglu, B.T. The clinical impact of chronopharmacology on current medicine. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Channel Type | Genes | Expression Change in BMAL1−/− Hearts | Functional Impact in Myocardial Cells | Reference |
|---|---|---|---|---|
| Sodium Channel | SCN5A | Decreased expression | Reduction of peak Ina by 30% in ventricular myocytes | [61] |
| Potassium Channel | KCND2, KCNH2 | Loss of expression | Reduction of Ikr peak by 50% in ventricular myocytes | [30] |
| KLF15, KCNIP2 | Loss of circadian expression | Reduced transcription levels of Kcni2; lack of Ito, f | [62] | |
| KCNIP2, KCNA5 | Decreased transcription levels | Unknown impact on ventricular myocytes | [51] | |
| KCNE1 | Increased expression | Increased susceptibility to atrial fibrillation | [63] | |
| Calcium Channel | CACNA1C | Weakened expression | Disrupted regulation on voltage and Ca2+ in cardiac myocytes | [64] |
| CASQ1, CASQ2 | Increased expression | Possible regulation on sarcoplasmic reticulum Ca2+ release and heart rate | [59,63] |
| Study | Animal Model | Major Findings | Implications |
|---|---|---|---|
| BMAL1 Knockout Studies | Mouse (Bmal1−/−) | Loss of circadian rhythmic oscillation in sodium (SCN5A) and potassium channels (KCND2, KCNH2), leading to impaired repolarization and increased arrhythmia susceptibility. | BMAL1 is crucial for maintaining circadian control of ion channels, impacting myocardial electrophysiology and arrhythmia risk. |
| Nav1.5 Channel Oscillation | Mouse | Circadian rhythmic oscillation in Nav1.5 expression affects myocardial depolarization; disrupted in Bmal1−/− models. | A direct role of BMAL1 in regulating sodium channel activity and the cardiac action potential. |
| Clock Gene Impact on Potassium Channels | Transgenic mice | BMAL1/CLOCK overexpression reduces potassium channel expression, affecting repolarization. | The role of circadian regulation in maintaining cardiac electrical stability. |
| L-Type Calcium Channel Regulation | Patch-clamp study in mouse myocardial cells | Clock/BMAL1 overexpression reduces L-type calcium current, leading to arrhythmia. | Demonstrates how circadian disruptions in calcium handling contribute to arrhythmic risk. |
| Epigenetic Impact of Doxorubicin (DOX) | Mouse model | DOX disrupts circadian homeostasis and alters protein acetylation, increasing cardiotoxicity. | Epigenetic modulation of circadian genes may offer therapeutic insights into reducing DOX-induced cardiotoxicity. |
| RORα Deficiency | Mouse (Rora−/−) | Exacerbates cardiac hypertrophy and dysfunction by impairing mitochondrial function. | RORα is critical for circadian regulation of mitochondrial biogenesis and cardiac function, with implications for heart failure therapy. |
| Histone Modification Effects | Mouse models | Changes in histone methylation (e.g., H3K4) impact ion channel expression and arrhythmogenesis. | Epigenetic mechanisms, like histone methylation, influence cardiac repolarization and arrhythmic susceptibility. |
| Ion Channel Type | Gene | Circadian Regulation | Effects of Circadian Disruption | Functional Consequence |
|---|---|---|---|---|
| Sodium (Na⁺) Channel | SCN5A | 24-h rhythmic oscillation in expression | Loss of circadian rhythmic expression; reduction in peak Ina by 30% | Impaired depolarization, increased arrhythmia susceptibility |
| Potassium (K⁺) Channel | KCND2, KCNH2 | 24-h rhythmic oscillation in expression | Loss of circadian rhythmic expression; reduction in peak Ikr by 50% | Prolonged action potential duration, impaired repolarization |
| KLF15, KCNIP2 | Circadian regulation by KLF15 | Reduced transcription levels; loss of Ito, f, prolonged ventricular AP duration | Impaired repolarization, prolonged AP duration | |
| KCNIP2, KCNA5 | Circadian expression regulated by BMAL1 | Decreased transcription levels | Potential impaired repolarization | |
| KCNE1 | Circadian regulation | Increased expression in BMAL1-altered mice; increased AF susceptibility | Increased susceptibility to atrial fibrillation | |
| Calcium (Ca2⁺) Channel | CACNA1C | Circadian rhythmic oscillation; regulated by CLOCK/BMAL1 | Weakened expression; reduced L-type calcium current | Abnormal calcium currents, triggering arrhythmias |
| CASQ1, CASQ2 | Circadian gene regulation by CLOCK/BMAL1 | Increased expression in BMAL1-altered mice | Dysregulated sarcoplasmic reticulum Ca2⁺ release, arrhythmogenic potential |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Li, S.; Zhang, H. Heart Failure and Arrhythmias: Circadian and Epigenetic Interplay in Myocardial Electrophysiology. Int. J. Mol. Sci. 2025, 26, 2728. https://doi.org/10.3390/ijms26062728
Zhu C, Li S, Zhang H. Heart Failure and Arrhythmias: Circadian and Epigenetic Interplay in Myocardial Electrophysiology. International Journal of Molecular Sciences. 2025; 26(6):2728. https://doi.org/10.3390/ijms26062728
Chicago/Turabian StyleZhu, Chen, Shuang Li, and Henggui Zhang. 2025. "Heart Failure and Arrhythmias: Circadian and Epigenetic Interplay in Myocardial Electrophysiology" International Journal of Molecular Sciences 26, no. 6: 2728. https://doi.org/10.3390/ijms26062728
APA StyleZhu, C., Li, S., & Zhang, H. (2025). Heart Failure and Arrhythmias: Circadian and Epigenetic Interplay in Myocardial Electrophysiology. International Journal of Molecular Sciences, 26(6), 2728. https://doi.org/10.3390/ijms26062728