
Tumor Microenvironment Dynamics of Triple-Negative Breast Cancer Under Radiation Therapy


Abstract
1. Introduction
1.1. Breast Cancer Biological and Molecular Subtypes
1.2. Biological Heterogeneity of TNBC and Its Impact on Clinical Outcomes
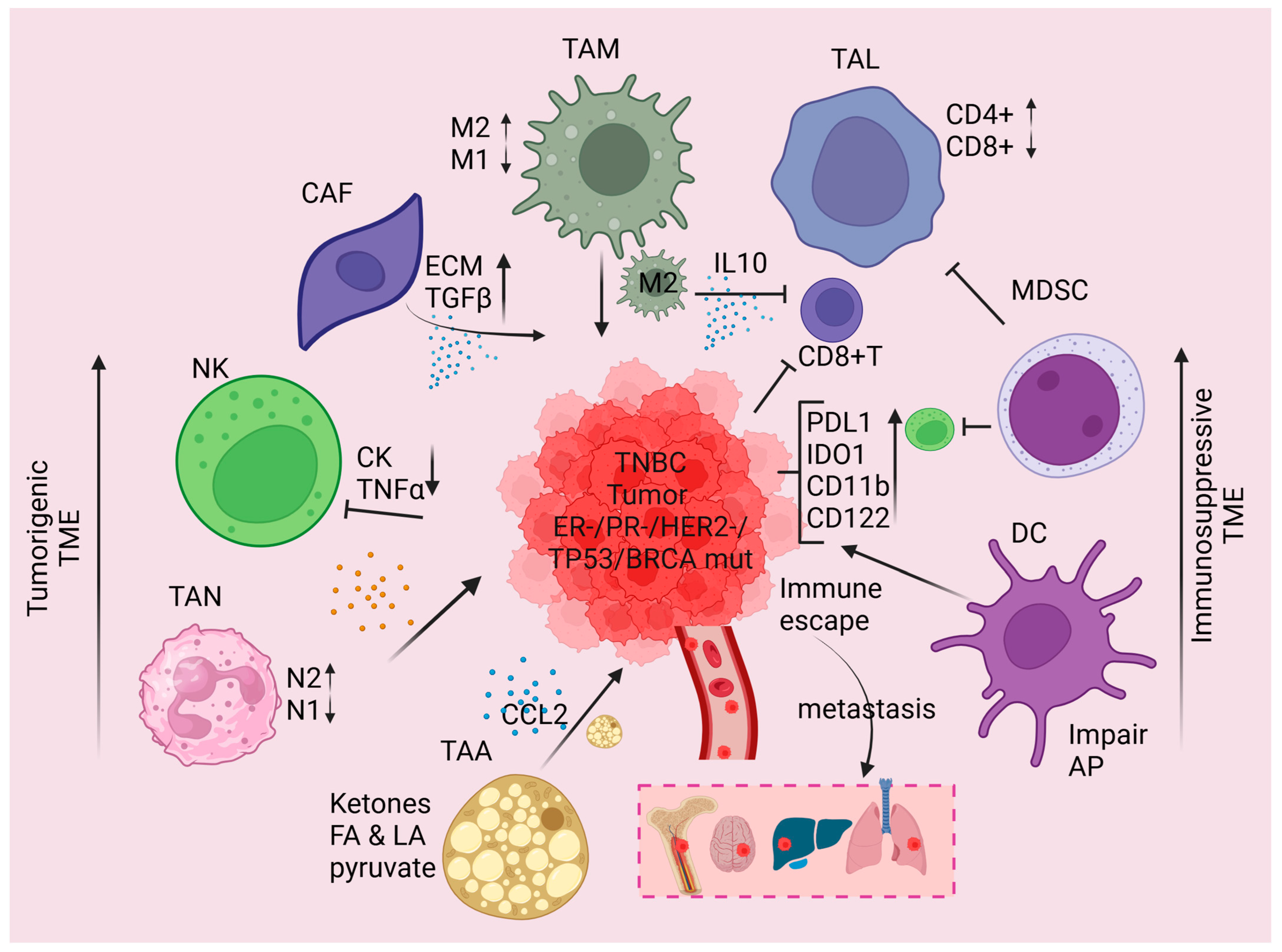
2. TNBC TME
2.1. Nature of TME
2.2. Interaction Between the TNBC Genomic Subclass and the TME
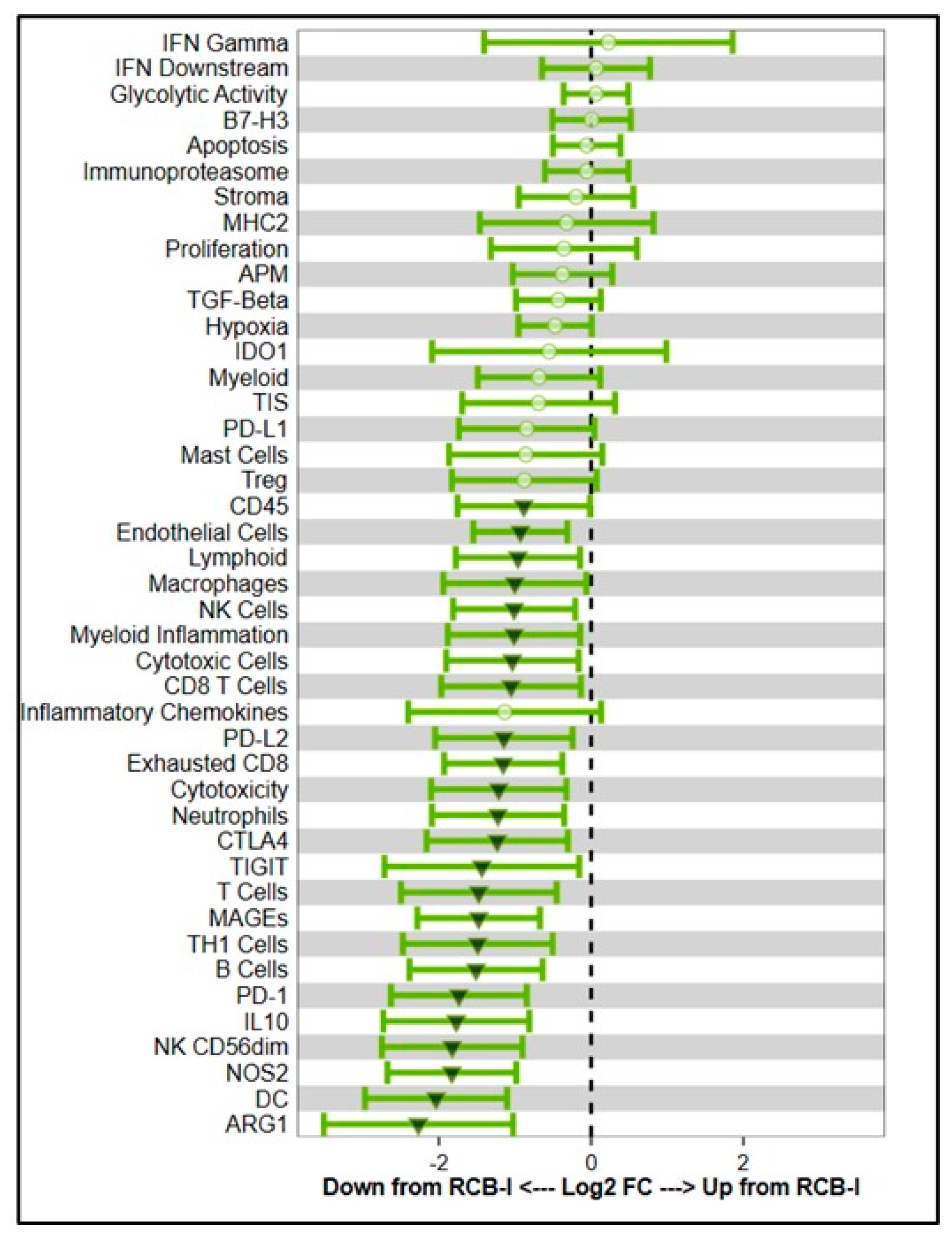
2.3. Immune Invasion Within the TNBC TME
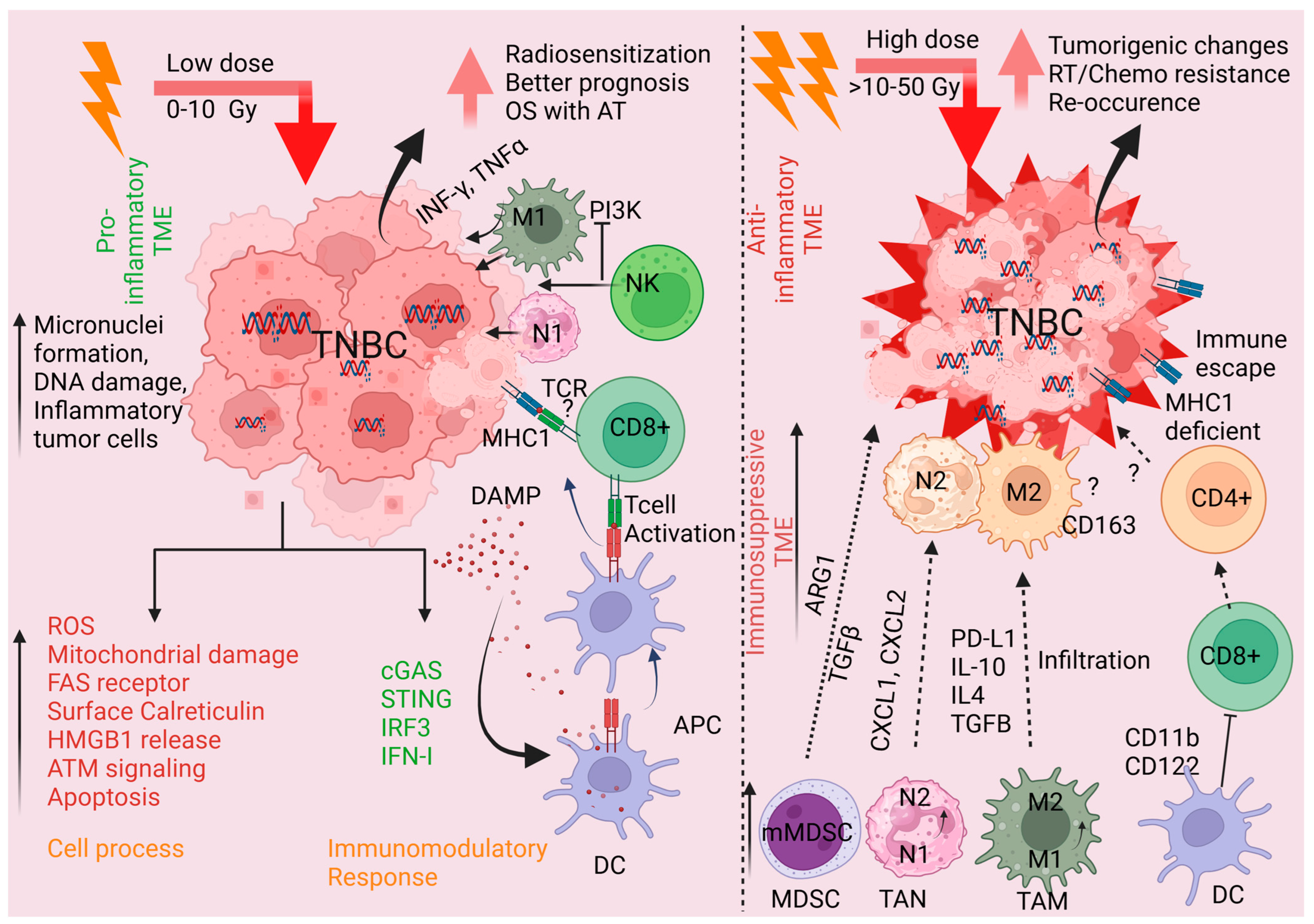
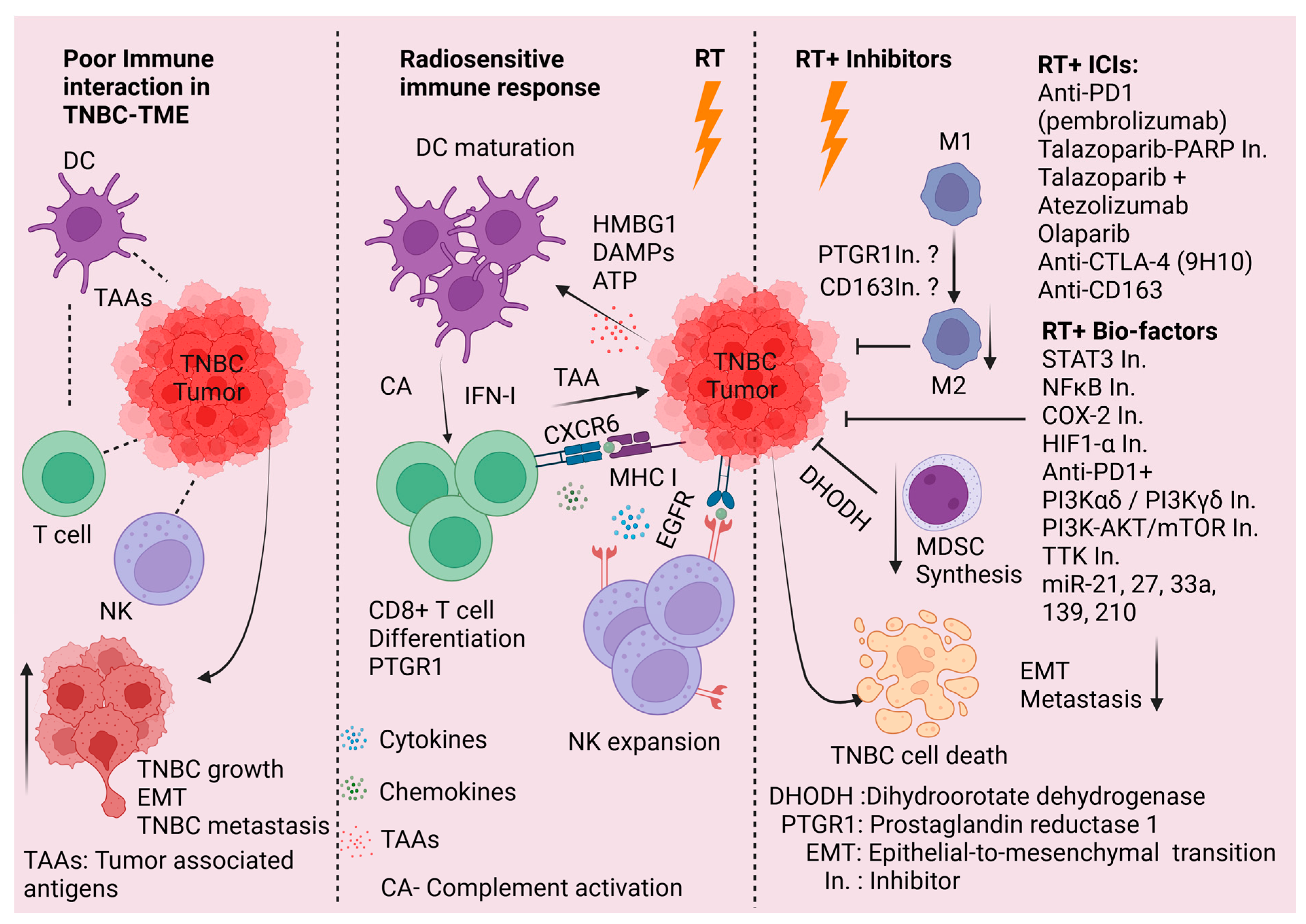
3. Radiation and the TME
3.1. Impact of RT on the TME
3.2. Potential Means of Therapeutically Exploiting RT-Mediated Impacts on TNBC TME
3.3. Radiosensitization of TNBC
3.4. Novel Clinical Trials Assessing Radiosensitization of TNBC
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, Y.; Gong, M.; Wang, Y.; Yang, Y.; Liu, S.; Zeng, Q. Global trends and forecasts of breast cancer incidence and deaths. Sci. Data 2023, 10, 334. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Sung, H.; Newman, L.A.; Freedman, R.A.; Smith, R.A.; Star, J.; Jemal, A.; Siegel, R.L. Breast cancer statistics 2024. CA Cancer J. Clin. 2024, 74, 477–495. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer-Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef] [PubMed]
- Aysola, K.; Desai, A.; Welch, C.; Xu, J.; Qin, Y.; Reddy, V.; Matthews, R.; Owens, C.; Okoli, J.; Beech, D.J.; et al. Triple Negative Breast Cancer—An Overview. Hered. Genet. 2013, 2013 (Suppl. 2), 001. [Google Scholar]
- Bergamino, M.A.; Lopez-Knowles, E.; Morani, G.; Tovey, H.; Kilburn, L.; Schuster, E.F.; Alataki, A.; Hills, M.; Xiao, H.; Holcombe, C.; et al. HER2-enriched subtype and novel molecular subgroups drive aromatase inhibitor resistance and an increased risk of relapse in early ER+/HER2+ breast cancer. EBioMedicine 2022, 83, 104205. [Google Scholar] [CrossRef]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. npj Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in Breast Cancer Survival by Molecular Subtypes in the United States. Cancer Epidemiol. Biomark. Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Y.; Song, B.; Yi, M.; Yan, Y.; Mei, Q.; Wu, K. Recent advances in targeted strategies for triple-negative breast cancer. J. Hematol. Oncol. 2023, 16, 100. [Google Scholar] [CrossRef]
- LeVee, A.; Wong, M.; Flores, S.; Ruel, N.; McArthur, H.; Waisman, J.; Mortimer, J. Impact of neoadjuvant pembrolizumab adherence on pathologic complete response in triple-negative breast cancer: A real-world analysis. Oncologist 2024, 29, 566–574. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef]
- Seal, M.D.; Chia, S.K. What Is the Difference Between Triple-Negative and Basal Breast Cancers? Cancer J. 2010, 16, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Pareja, F.; Weigelt, B.; Rakha, E.; Ellis, I.O.; Schnitt, S.J.; Reis-Filho, J.S. The Spectrum of Triple-Negative Breast Disease: High- and Low-Grade Lesions. Am. J. Pathol. 2017, 187, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; He, S. The Characteristics of Tumor Microenvironment in Triple Negative Breast Cancer. Cancer Manag. Res. 2022, 14, 1–17. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Li, R.Q.; Yan, L.; Zhang, L.; Ma, H.X.; Wang, H.W.; Bu, P.; Xi, Y.F.; Lian, J. Genomic characterization reveals distinct mutational landscapes and therapeutic implications between different molecular subtypes of triple-negative breast cancer. Sci. Rep. 2024, 14, 12386. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef]
- Tseng, Y.D.; Uno, H.; Hughes, M.E.; Niland, J.C.; Wong, Y.N.; Theriault, R.; Blitzblau, R.C.; Moy, B.; Breslin, T.; Edge, S.B.; et al. Biological Subtype Predicts Risk of Locoregional Recurrence After Mastectomy and Impact of Postmastectomy Radiation in a Large National Database. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 622–630. [Google Scholar] [CrossRef]
- Steward, L.T.; Gao, F.; Taylor, M.A.; Margenthaler, J.A. Impact of radiation therapy on survival in patients with triple-negative breast cancer. Oncol. Lett. 2014, 7, 548–552. [Google Scholar] [CrossRef]
- Sheva, K.; Roy Chowdhury, S.; Kravchenko-Balasha, N.; Meirovitz, A. Molecular Changes in Breast Cancer Induced by Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2024, 120, 465–481. [Google Scholar] [CrossRef]
- Pineiro Fernandez, J.; Luddy, K.A.; Harmon, C.; O’Farrelly, C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. Int. J. Mol. Sci. 2019, 20, 4131. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, Z.; Wang, Z.; Wu, P.; Qiu, F.; Huang, J. Prognostic and predictive value of tumor-infiltrating lymphocytes in breast cancer: A systematic review and meta-analysis. Clin. Transl. Oncol. 2016, 18, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Rey-Giraud, F.; Hafner, M.; Ries, C.H. In vitro generation of monocyte-derived macrophages under serum-free conditions improves their tumor promoting functions. PLoS ONE 2012, 7, e42656. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, K.; He, Q.; Gu, X.; Jiang, C.; Wu, J. Hypoxia signaling in cancer: Implications for therapeutic interventions. MedComm (2020) 2023, 4, e203. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125 Pt 23, 5591–5596. [Google Scholar] [CrossRef]
- Ahn, M.; Ali, A.; Seo, J.H. Mitochondrial regulation in the tumor microenvironment: Targeting mitochondria for immunotherapy. Front. Immunol. 2024, 15 Pt 23, 1453886. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, X.; Ye, J.; Li, H.; Hu, J.; Tan, Y.; Fang, Y.; Akbay, E.; Yu, F.; Weng, C.; et al. Systematic investigation of mitochondrial transfer between cancer cells and T cells at single-cell resolution. Cancer Cell 2023, 41, 1788–1802.e10. [Google Scholar] [CrossRef] [PubMed]
- Balta, E.; Wabnitz, G.H.; Samstag, Y. Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5736. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A. How tumors hijack macrophages for immune evasion. Nat. Cancer 2024, 5, 1134–1135. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothé, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef]
- Suntiparpluacha, M.; Chanthercrob, J.; Sa-Nguanraksa, D.; Sitthikornpaiboon, J.; Chaiboonchoe, A.; Kueanjinda, P.; Jinawath, N.; Sampattavanich, S. Retrospective study of transcriptomic profiling identifies Thai triple-negative breast cancer patients who may benefit from immune checkpoint and PARP inhibitors. PeerJ 2023, 11, e15350. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, W.; Shen, L.; Yao, Y.; Xia, W.; Ni, C. Tumor battlefield within inflamed, excluded or desert immune phenotypes: The mechanisms and strategies. Exp. Hematol. Oncol. 2024, 13, 80. [Google Scholar] [CrossRef]
- Kim, J.; Yu, D.; Kwon, Y.; Lee, K.S.; Sim, S.H.; Kong, S.Y.; Lee, E.S.; Park, I.H.; Park, C. Genomic Characteristics of Triple-Negative Breast Cancer Nominate Molecular Subtypes That Predict Chemotherapy Response. Mol. Cancer Res. 2020, 18, 253–263. [Google Scholar] [CrossRef]
- Thompson, K.J.; Leon-Ferre, R.A.; Sinnwell, J.P.; Zahrieh, D.M.; Suman, V.J.; Metzger, F.O.; Asad, S.; Stover, D.G.; Carey, L.; Sikov, W.M.; et al. Luminal androgen receptor breast cancer subtype and investigation of the microenvironment and neoadjuvant chemotherapy response. NAR Cancer 2022, 4, zcac018. [Google Scholar] [CrossRef]
- Rodriguez-Bautista, R.; Caro-Sanchez, C.H.; Cabrera-Galeana, P.; Alanis-Funes, G.J.; Gutierrez-Millan, E.; Avila-Rios, S.; Matias-Florentino, M.; Reyes-Teran, G.; Diaz-Chavez, J.; Villarreal-Garza, C.; et al. Immune Milieu and Genomic Alterations Set the Triple-Negative Breast Cancer Immunomodulatory Subtype Tumor Behavior. Cancers 2021, 13, 6256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Xu, X.; Li, X.; Guan, W.; Meng, T.; Xu, G. Transcriptome-Based Network Analysis Unveils Eight Immune-Related Genes as Molecular Signatures in the Immunomodulatory Subtype of Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Tsuda, H.; Shimizu, C.; Yamamoto, S.; Shibata, T.; Yamamoto, H.; Hirata, T.; Yonemori, K.; Ando, M.; Tamura, K.; et al. Tumor-infiltrating lymphocytes are correlated with response to neoadjuvant chemotherapy in triple-negative breast cancer. Breast Cancer Res. Treat. 2012, 132, 793–805. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, C.; Zhang, Q.; Ye, J.; Wang, F.; Zhang, Y.; Hunborg, P.; Varvares, M.A.; Hoft, D.F.; Hsueh, E.C.; et al. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 2015, 6, 17462–17478. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, M.; Li, C.; Liu, X.; Zhao, J.; Ma, H.; Zhang, S.; Qu, J. High dose Vitamin C inhibits PD-L1 by ROS-pSTAT3 signal pathway and enhances T cell function in TNBC. Int. Immunopharmacol. 2024, 126, 111321. [Google Scholar] [CrossRef]
- Saravanan, R.; Balasubramanian, V.; Sundaram, S.; Dev, B.; Vittalraj, P.; Pitani, R.S.; Shanmugasundaram, G.; Rayala, S.K.; Venkatraman, G. Expression of cell surface zinc transporter LIV1 in triple negative breast cancer is an indicator of poor prognosis and therapy failure. J. Cell. Physiol. 2024, 239, e31203. [Google Scholar] [CrossRef]
- Harris, R.J.; Cheung, A.; Ng, J.C.F.; Laddach, R.; Chenoweth, A.M.; Crescioli, S.; Fittall, M.; Dominguez-Rodriguez, D.; Roberts, J.; Levi, D.; et al. Tumor-Infiltrating B Lymphocyte Profiling Identifies IgG-Biased, Clonally Expanded Prognostic Phenotypes in Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 4290–4304. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, C.; Xu, T.; Lan, Y.; Lin, H.; Wu, X.; Li, X. Single-cell and bulk RNA sequencing analysis of B cell marker genes in TNBC TME landscape and immunotherapy. Front. Immunol. 2023, 14, 1245514. [Google Scholar] [CrossRef]
- Tenggara, J.B.; Rachman, A.; Prihartono, J.; Rachmadi, L.; Panigoro, S.S.; Heriyanto, D.S.; Sutandyo, N.; Nasution, I.R.; Rahadiati, F.B.; Steven, R.; et al. The relationship between high ratios of CD4/FOXP3 and CD8/CD163 and the improved survivability of metastatic triple-negative breast cancer patients: A multicenter cohort study. BMC Res. Notes 2024, 17, 44. [Google Scholar] [CrossRef]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Leon, M.L.; Jimenez-Cortegana, C.; Silva Romeiro, S.; Garnacho, C.; de la Cruz-Merino, L.; Garcia-Dominguez, D.J.; Hontecillas-Prieto, L.; Sanchez-Margalet, V. Defining the Emergence of New Immunotherapy Approaches in Breast Cancer: Role of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2023, 24, 5208. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Ghosh, S.; Huang, J.; Inkman, M.; Zhang, J.; Thotala, S.; Tikhonova, E.; Miheecheva, N.; Frenkel, F.; Ataullakhanov, R.; Wang, X.; et al. Radiation-induced circulating myeloid-derived suppressor cells induce systemic lymphopenia after chemoradiotherapy in patients with glioblastoma. Sci. Transl. Med. 2023, 15, eabn6758. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, eaay6017. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Cha, Y.J.; Koo, J.S. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells 2020, 9, 1785. [Google Scholar] [CrossRef]
- Colligan, S.H.; Amitrano, A.M.; Zollo, R.A.; Peresie, J.; Kramer, E.D.; Morreale, B.; Barbi, J.; Singh, P.K.; Yu, H.; Wang, J.; et al. Inhibiting the biogenesis of myeloid-derived suppressor cells enhances immunotherapy efficacy against mammary tumor progression. J. Clin. Investig. 2022, 132, e158661. [Google Scholar] [CrossRef]
- Gebremeskel, S.; Clattenburg, D.R.; Slauenwhite, D.; Lobert, L.; Johnston, B. Natural killer T cell activation overcomes immunosuppression to enhance clearance of postsurgical breast cancer metastasis in mice. Oncoimmunology 2015, 4, e995562. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhao, T.; Luo, R.; Qiu, R.; Li, Z. Tumor-Associated Macrophages: Key Players in Triple-Negative Breast Cancer. Front. Oncol. 2022, 12, 772615. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.-Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schröder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 macrophages and their overlaps—Myth or reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef]
- Stavrou, M.; Constantinidou, A. Tumor associated macrophages in breast cancer progression: Implications and clinical relevance. Front. Immunol. 2024, 15, 1441820. [Google Scholar] [CrossRef]
- Hirano, R.; Okamoto, K.; Shinke, M.; Sato, M.; Watanabe, S.; Watanabe, H.; Kondoh, G.; Kadonosono, T.; Kizaka-Kondoh, S. Tissue-resident macrophages are major tumor-associated macrophage resources, contributing to early TNBC development, recurrence, and metastases. Commun. Biol. 2023, 6, 144. [Google Scholar] [CrossRef]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Chen, X.; Lin, H. Expression of Tumor-Related Macrophages and Cytokines After Surgery of Triple-Negative Breast Cancer Patients and its Implications. Med. Sci. Monit. 2016, 22, 115–120. [Google Scholar] [CrossRef]
- Matkowski, R.; Gisterek, I.; Halon, A.; Lacko, A.; Szewczyk, K.; Staszek, U.; Pudelko, M.; Szynglarewicz, B.; Szelachowska, J.; Zolnierek, A.; et al. The prognostic role of tumor-infiltrating CD4 and CD8 T lymphocytes in breast cancer. Anticancer Res. 2009, 29, 2445–2451. [Google Scholar]
- Jaaskelainen, M.M.; Tumelius, R.; Hamalainen, K.; Rilla, K.; Oikari, S.; Ronka, A.; Selander, T.; Mannermaa, A.; Tiainen, S.; Auvinen, P. High Numbers of CD163+ Tumor-Associated Macrophages Predict Poor Prognosis in HER2+ Breast Cancer. Cancers 2024, 16, 643. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Hwang, I.; Kang, S.H.; Shin, H.C.; Kwon, S.Y. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J. Breast Cancer 2019, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Omilian, A.R.; Cannioto, R.; Mendicino, L.; Stein, L.; Bshara, W.; Qin, B.; Bandera, E.V.; Zeinomar, N.; Abrams, S.I.; Hong, C.C.; et al. CD163(+) macrophages in the triple-negative breast tumor microenvironment are associated with improved survival in the Women’s Circle of Health Study and the Women’s Circle of Health Follow-Up Study. Breast Cancer Res. 2024, 26, 75. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, M.; Yin, J.; Li, P.; Zeng, S.; Zheng, G.; He, Z.; Liu, H.; Wang, Q.; Zhang, F.; et al. Tumor-associated macrophages promote epithelial–mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/β-catenin signaling. Cell Commun. Signal. 2022, 20, 92. [Google Scholar] [CrossRef]
- Deng, X.X.; Jiao, Y.N.; Hao, H.F.; Xue, D.; Bai, C.C.; Han, S.Y. Taraxacum mongolicum extract inhibited malignant phenotype of triple-negative breast cancer cells in tumor-associated macrophages microenvironment through suppressing IL-10 / STAT3 / PD-L1 signaling pathways. J. Ethnopharmacol. 2021, 274, 113978. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Magesh, P.; Thankachan, S.; Venkatesh, T.; Suresh, P.S. Breast cancer fibroblasts and cross-talk. Clin. Chim. Acta 2021, 521, 158–169. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Huang, Y.; Ji, S.; Shao, G.; Feng, S.; Chen, D.; Zhao, K.; Wang, Z.; Wu, A. Cancer-Associated Fibroblasts Autophagy Enhances Progression of Triple-Negative Breast Cancer Cells. Med. Sci. Monit. 2017, 23, 3904–3912. [Google Scholar] [CrossRef]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.T.; Elloumi, F.; Roman-Perez, E.; Rein, J.; Stewart, D.A.; Harrell, J.C.; Perou, C.M.; Troester, M.A. Interactions with fibroblasts are distinct in Basal-like and luminal breast cancers. Mol. Cancer Res. 2011, 9, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Allaoui, R.; Bergenfelz, C.; Mohlin, S.; Hagerling, C.; Salari, K.; Werb, Z.; Anderson, R.L.; Ethier, S.P.; Jirstrom, K.; Pahlman, S.; et al. Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat. Commun. 2016, 7, 13050. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, H.; Shen, X.; Jin, W.; Wang, X.; Zhou, Z. Characterization of cancer-associated fibroblasts (CAFs) and development of a CAF-based risk model for triple-negative breast cancer. Cancer Cell Int. 2023, 23, 294. [Google Scholar] [CrossRef]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef]
- Rymaszewski, A.L.; Tate, E.; Yimbesalu, J.P.; Gelman, A.E.; Jarzembowski, J.A.; Zhang, H.; Pritchard, K.A., Jr.; Vikis, H.G. The role of neutrophil myeloperoxidase in models of lung tumor development. Cancers 2014, 6, 1111–1127. [Google Scholar] [CrossRef]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast cancer cells stimulate neutrophils to produce oncostatin M: Potential implications for tumor progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef]
- Liang, B.; Yuan, Y.; Jiang, Q.; Ma, T.; Liu, X.; Li, Y. How neutrophils shape the immune response of triple-negative breast cancer: Novel therapeutic strategies targeting neutrophil extracellular traps. Biomed. Pharmacother. 2024, 178, 117211. [Google Scholar] [CrossRef]
- SenGupta, S.; Hein, L.E.; Xu, Y.; Zhang, J.; Konwerski, J.R.; Li, Y.; Johnson, C.; Cai, D.; Smith, J.L.; Parent, C.A. Triple-Negative Breast Cancer Cells Recruit Neutrophils by Secreting TGF-beta and CXCR2 Ligands. Front. Immunol. 2021, 12, 659996. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Julia, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Mathias, C.; Kozak, V.N.; Magno, J.M.; Baal, S.C.S.; Dos Santos, V.H.A.; Ribeiro, E.; Gradia, D.F.; Castro, M.A.A.; Carvalho de Oliveira, J. PD-1/PD-L1 Inhibitors Response in Triple-Negative Breast Cancer: Can Long Noncoding RNAs Be Associated? Cancers 2023, 15, 4682. [Google Scholar] [CrossRef]
- Xu, Y.; Carrascosa, L.C.; Yeung, Y.A.; Chu, M.L.; Yang, W.; Djuretic, I.; Pappas, D.C.; Zeytounian, J.; Ge, Z.; de Ruiter, V.; et al. An Engineered IL15 Cytokine Mutein Fused to an Anti-PD1 Improves Intratumoral T-cell Function and Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 1141–1157. [Google Scholar] [CrossRef]
- Ma, S.; Han, J.; Li, Z.; Xiao, S.; Zhang, J.; Yan, J.; Tang, T.; Barr, T.; Kraft, A.S.; Caligiuri, M.A.; et al. An XBP1s-PIM-2 positive feedback loop controls IL-15-mediated survival of natural killer cells. Sci. Immunol. 2023, 8, eabn7993. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Tian, J.; Hao, Y.; Ma, X.; Zhou, Y.; Feng, L. Engineered CAR-NK Cells with Tolerance to H2O2 and Hypoxia Can Suppress Postoperative Relapse of Triple-Negative Breast Cancers. Cancer Immunol. Res. 2024, 12, 1574–1588. [Google Scholar] [CrossRef]
- Li, T.; Niu, M.; Zhang, W.; Qin, S.; Zhou, J.; Yi, M. CAR-NK cells for cancer immunotherapy: Recent advances and future directions. Front. Immunol. 2024, 15, 1361194. [Google Scholar] [CrossRef]
- Wlodarczyk, M.; Pyrzynska, B. CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer. Cancers 2022, 15, 117. [Google Scholar] [CrossRef]
- Wu, C.; Dong, S.; Huang, R.; Chen, X. Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy. Cancers 2023, 15, 726. [Google Scholar] [CrossRef]
- Fujisaki, K.; Fujimoto, H.; Sangai, T.; Nagashima, T.; Sakakibara, M.; Shiina, N.; Kuroda, M.; Aoyagi, Y.; Miyazaki, M. Cancer-mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res. Treat. 2015, 150, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef] [PubMed]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun. 2011, 411, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Luo, N.; Ma, L.; Ma, N.; Wei, J.; Zhang, H.; Jin, W.; Li, Y.; Shi, J.; Xiong, Y. Hesperidin PLGA nanoparticles potentiate the efficacy of aPD-1 in treating triple negative breast cancer by regulating CCL2 and ADPN expression in cancer-associated adipocytes. Int. Immunopharmacol. 2024, 140, 112759. [Google Scholar] [CrossRef]
- Huang, R.; Wang, Z.; Hong, J.; Wu, J.; Huang, O.; He, J.; Chen, W.; Li, Y.; Chen, X.; Shen, K. Targeting cancer-associated adipocyte-derived CXCL8 inhibits triple-negative breast cancer progression and enhances the efficacy of anti-PD-1 immunotherapy. Cell Death Dis. 2023, 14, 703. [Google Scholar] [CrossRef]
- Charpentier, M.; Spada, S.; Van Nest, S.J.; Demaria, S. Radiation therapy-induced remodeling of the tumor immune microenvironment. Semin. Cancer Biol. 2022, 86, 737–747. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Citrin, D.E. Recent Developments in Radiotherapy. N. Engl. J. Med. 2017, 377, 2200–2201. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Stueber, T.N.; Diessner, J.; Bartmann, C.; Leinert, E.; Janni, W.; Herr, D.; Kreienberg, R.; Woeckel, A.; Wischnewsky, M. Effect of adjuvant radiotherapy in elderly patients with breast cancer. PLoS ONE 2020, 15, e0229518. [Google Scholar] [CrossRef]
- Dosani, M.; Schrader, K.A.; Nichol, A.; Sun, S.; Shenkier, T.; Lohn, Z.; Aubertin, G.; Tyldesley, S. Severe Late Toxicity After Adjuvant Breast Radiotherapy in a Patient with a Germline Ataxia Telangiectasia Mutated Gene: Future Treatment Decisions. Cureus 2017, 9, e1458. [Google Scholar] [CrossRef]
- Reislander, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef]
- Ruckert, M.; Flohr, A.S.; Hecht, M.; Gaipl, U.S. Radiotherapy and the immune system: More than just immune suppression. Stem Cells 2021, 39, 1155–1165. [Google Scholar] [CrossRef]
- Dutt, S.; Ahmed, M.M.; Loo, B.W., Jr.; Strober, S. Novel Radiation Therapy Paradigms and Immunomodulation: Heresies and Hope. Semin. Radiat. Oncol. 2020, 30, 194–200. [Google Scholar] [CrossRef]
- Fultang, N.; Li, X.; Li, T.; Chen, Y.H. Myeloid-Derived Suppressor Cell Differentiation in Cancer: Transcriptional Regulators and Enhanceosome-Mediated Mechanisms. Front. Immunol. 2020, 11, 619253. [Google Scholar] [CrossRef]
- Weber, R.; Riester, Z.; Huser, L.; Sticht, C.; Siebenmorgen, A.; Groth, C.; Hu, X.; Altevogt, P.; Utikal, J.S.; Umansky, V. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J. Immunother. Cancer 2020, 8, e000949. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Johanns, T.M.; Chheda, M.G.; Liu, E.; Butt, O.; Abraham, C.; Badiyan, S.; Huang, Y.; DeNardo, D.; Kim, A.H.; et al. A pilot phase Ib study to evaluate tadalafil to overcome immunosuppression during chemoradiotherapy for IDH-wild-type glioblastoma. Neurooncol. Adv. 2023, 5, vdad088. [Google Scholar] [CrossRef] [PubMed]
- Rezapour, M.; Walker, S.J.; Ornelles, D.A.; Niazi, M.K.K.; McNutt, P.M.; Atala, A.; Gurcan, M.N. A comparative analysis of RNA-Seq and NanoString technologies in deciphering viral infection response in upper airway lung organoids. Front. Genet. 2024, 15, 1327984. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef]
- Chamoto, K.; Al-Habsi, M.; Honjo, T. Role of PD-1 in Immunity and Diseases. Curr. Top. Microbiol. Immunol. 2017, 410, 75–97. [Google Scholar]
- Sceneay, J.; Goreczny, G.J.; Wilson, K.; Morrow, S.; DeCristo, M.J.; Ubellacker, J.M.; Qin, Y.; Laszewski, T.; Stover, D.G.; Barrera, V.; et al. Interferon Signaling Is Diminished with Age and Is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 1208–1227. [Google Scholar] [CrossRef]
- He, S.; Cheng, J.; Sun, L.; Wang, Y.; Wang, C.; Liu, X.; Zhang, Z.; Zhao, M.; Luo, Y.; Tian, L.; et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018, 9, 648. [Google Scholar] [CrossRef]
- He, M.Y.; Rancoule, C.; Rehailia-Blanchard, A.; Espenel, S.; Trone, J.-C.; Bernichon, E.; Guillaume, E.; Vallard, A.; Magné, N. Radiotherapy in triple-negative breast cancer: Current situation and upcoming strategies. Crit. Rev. Oncol. Hematol. 2018, 131, 96–101. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and Immunogenic Cell Death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef]
- Shang, L.; Zhong, Y.; Yao, Y.; Liu, C.; Wang, L.; Zhang, W.; Liu, J.; Wang, X.; Sun, C. Subverted macrophages in the triple-negative breast cancer ecosystem. Biomed. Pharmacother. 2023, 166, 115414. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Hehlgans, S.; Rödel, F.; Gaipl, U.S. Modulation of inflammation by low and high doses of ionizing radiation: Implications for benign and malign diseases. Cancer Lett. 2015, 368, 230–237. [Google Scholar] [CrossRef]
- Leblond, M.M.; Peres, E.A.; Helaine, C.; Gerault, A.N.; Moulin, D.; Anfray, C.; Divoux, D.; Petit, E.; Bernaudin, M.; Valable, S. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget 2017, 8, 72597–72612. [Google Scholar] [CrossRef] [PubMed]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, W.; Hu, S.; Jia, B.; Tuo, B.; Sun, H.; Wang, Q.; Liu, Y.; Sun, Z. Radiotherapy remodels the tumor microenvironment for enhancing immunotherapeutic sensitivity. Cell Death Dis. 2023, 14, 679. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Meng, J.; Yang, Y.; Lv, J.; Lv, H.; Zhao, X.; Zhang, L.; Shi, W.; Yang, Z.; Mei, X.; Chen, X.; et al. CXCR6 expression correlates with radiotherapy response and immune context in triple-negative breast cancer-experimental studies. Int. J. Surg. 2024, 110, 4695–4707. [Google Scholar] [CrossRef]
- Huang, F.; Wang, F.; Hu, Q.; Li, Y.; Jiang, D. PTGR1-mediated immune evasion mechanisms in late-stage triple-negative breast cancer: Mechanisms of M2 macrophage infiltration and CD8(+) T cell suppression. Apoptosis 2024, 29, 2002–2024. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, M.; Shi, B.; Di, S.; Sun, R.; Jiang, H.; Li, Z. Radiation enhances the efficacy of EGFR-targeted CAR-T cells against triple-negative breast cancer by activating NF-kappaB/Icam1 signaling. Mol. Ther. 2022, 30, 3379–3393. [Google Scholar] [CrossRef]
- Kim, K.W.; Jeong, J.U.; Lee, K.H.; Uong, T.N.T.; Rhee, J.H.; Ahn, S.J.; Kim, S.K.; Cho, D.; Quang Nguyen, H.P.; Pham, C.T.; et al. Combined NK Cell Therapy and Radiation Therapy Exhibit Long-Term Therapeutic and Antimetastatic Effects in a Human Triple Negative Breast Cancer Model. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 115–125. [Google Scholar] [CrossRef]
- Chang, W.I.; Han, M.G.; Kang, M.H.; Park, J.M.; Kim, E.E.; Bae, J.; Ahn, S.; Kim, I.A. PI3Kalphadelta Inhibitor Combined With Radiation Enhances the Antitumor Immune Effect of Anti-PD1 in a Syngeneic Murine Triple-Negative Breast Cancer Model. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.N.; Lee, E.; Kwon, Y.J.; Gim, J.A.; Kim, T.J.; Kim, J.S. PI3Kdelta/gamma inhibitor BR101801 extrinsically potentiates effector CD8(+) T cell-dependent antitumor immunity and abscopal effect after local irradiation. J. Immunother. Cancer 2022, 10, e003762. [Google Scholar] [CrossRef] [PubMed]
- Song, H.N.; Jin, H.; Kim, J.H.; Ha, I.B.; Kang, K.M.; Choi, H.S.; Jeong, H.J.; Kim, M.Y.; Kim, H.J.; Jeong, B.K. Abscopal Effect of Radiotherapy Enhanced with Immune Checkpoint Inhibitors of Triple Negative Breast Cancer in 4T1 Mammary Carcinoma Model. Int. J. Mol. Sci. 2021, 22, 10476. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11 Pt 1, 728–734. [Google Scholar] [CrossRef]
- Clark, C.A.; Yang, E.S. Harnessing DNA Repair Defects to Augment Immune-Based Therapies in Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 703802. [Google Scholar] [CrossRef]
- Guo, S.; Yao, Y.; Tang, Y.; Xin, Z.; Wu, D.; Ni, C.; Huang, J.; Wei, Q.; Zhang, T. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct. Target. Ther. 2023, 8, 205. [Google Scholar] [CrossRef]
- Zhang, B.; Hu, M.; Ma, Q.; Li, K.; Li, X.; He, X.; Shu, P.; Chen, Y.; Gao, G.; Qin, D.; et al. Optimized CAR-T therapy based on spatiotemporal changes and chemotactic mechanisms of MDSCs induced by hypofractionated radiotherapy. Mol. Ther. 2023, 31, 2105–2119. [Google Scholar] [CrossRef]
- Vito, A.; Rathmann, S.; Mercanti, N.; El-Sayes, N.; Mossman, K.; Valliant, J. Combined Radionuclide Therapy and Immunotherapy for Treatment of Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4843. [Google Scholar] [CrossRef]
- Marni, R.; Malla, M.; Chakraborty, A.; Voonna, M.K.; Bhattacharyya, P.S.; Kgk, D.; Malla, R.R. Combination of ionizing radiation and 2-thio-6-azauridine induces cell death in radioresistant triple negative breast cancer cells by downregulating CD151 expression. Cancer Chemother. Pharmacol. 2024, 94, 685–706. [Google Scholar] [CrossRef]
- Wang, D.; Lin, S.; Li, T.; Yang, X.; Zhong, X.; Chen, Q.; Jiang, G.; Li, C. Cancer cell membrane-coated siRNA-Decorated Au/MnO2 nanosensitizers for synergistically enhanced radio-immunotherapy of breast cancer. Mater. Today Bio 2024, 29, 101275. [Google Scholar] [CrossRef]
- Loap, P.; Loirat, D.; Berger, F.; Ricci, F.; Vincent-Salomon, A.; Ezzili, C.; Mosseri, V.; Fourquet, A.; Ezzalfani, M.; Kirova, Y. Combination of Olaparib and Radiation Therapy for Triple Negative Breast Cancer: Preliminary Results of the RADIOPARP Phase 1 Trial. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Zellars, R.C.; Lange, J.R.; Habibi, M.; Fetting, J.H.; Stearns, V. Preoperative PARPi and irradiation (POPI) for women with an incomplete response to neoadjuvant chemotherapy (NAC) for breast cancer: A phase I trial. J. Clin. Oncol. 2014, 32 (Suppl. S15), TPS1142. [Google Scholar] [CrossRef]
- Ho, A.Y.; Barker, C.A.; Arnold, B.B.; Powell, S.N.; Hu, Z.I.; Gucalp, A.; Lebron-Zapata, L.; Wen, H.Y.; Kallman, C.; D’Agnolo, A.; et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 2020, 126, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.A.; Kalinsky, K.; Stringer-Reasor, E.; Elkhanany, A.; Lin, J.; Schuster, D.M.; Friend, S.; Switchenko, J.; Bhave, M. Abstract OT2-10-03: HCRN BRE 19-433: A Multi-institutional Phase II Study to Evaluate Efficacy and Safety of TAlazoparib, Radiotherapy and Atezolizumab in gBRCA 1/2 negative Patients with PD-L1+ Metastatic Triple Negative Breast Cancer (TARA). Cancer Res. 2023, 83 (Suppl. 5), OT2-10-03. [Google Scholar] [CrossRef]
- Alavimanesh, S.; Nayerain Jazi, N.; Choubani, M.; Saeidi, F.; Afkhami, H.; Yarahmadi, A.; Ronaghi, H.; Khani, P.; Modarressi, M.H. Cellular senescence in the tumor with a bone niche microenvironment: Friend or foe? Clin. Exp. Med. 2025, 25, 44. [Google Scholar] [CrossRef]
- Shupp, A.B.; Kolb, A.D.; Mukhopadhyay, D.; Bussard, K.M. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin. Transl. Radiat. Oncol. 2020, 22, 90–97. [Google Scholar] [CrossRef]
- Zhao, D.; Mo, Y.; Neganova, M.E.; Aleksandrova, Y.; Tse, E.; Chubarev, V.N.; Fan, R.; Sukocheva, O.A.; Liu, J. Dual effects of radiotherapy on tumor microenvironment and its contribution towards the development of resistance to immunotherapy in gastrointestinal and thoracic cancers. Front. Cell Dev. Biol. 2023, 11, 1266537. [Google Scholar] [CrossRef]
- Golebiewska, A.; Fields, R.C. Advancing preclinical cancer models to assess clinically relevant outcomes. BMC Cancer 2023, 23, 230. [Google Scholar] [CrossRef]
- Miserocchi, G.; Bocchini, M.; Cortesi, M.; Arienti, C.; De Vita, A.; Liverani, C.; Mercatali, L.; Bravaccini, S.; Ulivi, P.; Zanoni, M. Combining preclinical tools and models to unravel tumor complexity: Jump into the next dimension. Front. Immunol. 2023, 14, 1171141. [Google Scholar] [CrossRef] [PubMed]
- Stribbling, S.M.; Beach, C.; Ryan, A.J. Orthotopic and metastatic tumour models in preclinical cancer research. Pharmacol. Ther. 2024, 257, 108631. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Niture, S.; Jaboin, J.; Seneviratne, D. Abstract B007: CD163+ tumor-associated macrophage evasion contributes radiation resistance and poor prognosis in estrogen receptor-negative breast cancer. Clin. Cancer Res. 2025, 31 (Suppl. S2), B007. [Google Scholar] [CrossRef]
- Li, G.; Lin, X.; Wang, X.; Cai, L.; Liu, J.; Zhu, Y.; Fu, Z. Enhancing radiosensitivity in triple-negative breast cancer through targeting ELOB. Breast Cancer 2024, 31, 426–439. [Google Scholar] [CrossRef]
- Wen, Y.; Dai, G.; Wang, L.; Fu, K.; Zuo, S. Silencing of XRCC4 increases radiosensitivity of triple-negative breast cancer cells. Biosci. Rep. 2019, 39, BSR20180893. [Google Scholar] [CrossRef]
- Parsyan, A.; Cruickshank, J.; Hodgson, K.; Wakeham, D.; Pellizzari, S.; Bhat, V.; Cescon, D.W. Anticancer effects of radiation therapy combined with Polo-Like Kinase 4 (PLK4) inhibitor CFI-400945 in triple negative breast cancer. Breast 2021, 58, 6–9. [Google Scholar] [CrossRef]
- Pellizzari, S.; Bhat, V.; Athwal, H.; Cescon, D.W.; Allan, A.L.; Parsyan, A. PLK4 as a potential target to enhance radiosensitivity in triple-negative breast cancer. Radiat. Oncol. 2024, 19, 24. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Chen, W.-M.; Hassan, O.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Green, M.D.; Davis, A.J.; Speers, C.; et al. BUB1 regulates non-homologous end joining pathway to mediate radioresistance in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2024, 43, 163. [Google Scholar] [CrossRef]
- Chandler, B.C.; Moubadder, L.; Ritter, C.L.; Liu, M.; Cameron, M.; Wilder-Romans, K.; Zhang, A.; Pesch, A.M.; Michmerhuizen, A.R.; Hirsh, N.; et al. TTK inhibition radiosensitizes basal-like breast cancer through impaired homologous recombination. J. Clin. Investig. 2020, 130, 958–973. [Google Scholar] [CrossRef]
- Zetrini, A.E.; Abbasi, A.Z.; He, C.; Lip, H.; Alradwan, I.; Rauth, A.M.; Henderson, J.T.; Wu, X.Y. Targeting DNA damage repair mechanism by using RAD50-silencing siRNA nanoparticles to enhance radiotherapy in triple negative breast cancer. Mater. Today Bio 2024, 28, 101206. [Google Scholar] [CrossRef]
- Heravi, M.; Kumala, S.; Rachid, Z.; Jean-Claude, B.J.; Radzioch, D.; Muanza, T.M. ZRBA1, a Mixed EGFR/DNA Targeting Molecule, Potentiates Radiation Response Through Delayed DNA Damage Repair Process in a Triple Negative Breast Cancer Model. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Speers, C.; Zhao, S.G.; Kothari, V.; Santola, A.; Liu, M.; Wilder-Romans, K.; Evans, J.; Batra, N.; Bartelink, H.; Hayes, D.F.; et al. Maternal Embryonic Leucine Zipper Kinase (MELK) as a Novel Mediator and Biomarker of Radioresistance in Human Breast Cancer. Clin. Cancer Res. 2016, 22, 5864–5875. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Liu, N.; Zheng, D.; Du, J.; Wang, K. MicroRNA-206 inhibits metastasis of triple-negative breast cancer by targeting transmembrane 4 L6 family member 1. Cancer Manag. Res. 2019, 11, 6755–6764. [Google Scholar] [CrossRef] [PubMed]
- Maskey, N.; Li, D.; Xu, H.; Song, H.; Wu, C.; Hua, K.; Song, J.; Fang, L. MicroRNA-340 inhibits invasion and metastasis by downregulating ROCK1 in breast cancer cells. Oncol. Lett. 2017, 14, 2261–2267. [Google Scholar] [CrossRef]
- Shi, P.; Chen, C.; Li, X.; Wei, Z.; Liu, Z.; Liu, Y. MicroRNA-124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol. Med. Rep. 2019, 19, 3667–3675. [Google Scholar] [CrossRef]
- Saatci, O.; Kaymak, A.; Raza, U.; Ersan, P.G.; Akbulut, O.; Banister, C.E.; Sikirzhytski, V.; Tokat, U.M.; Aykut, G.; Ansari, S.A.; et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat. Commun. 2020, 11, 2416. [Google Scholar] [CrossRef]
- Fang, H.; Xie, J.; Zhang, M.; Zhao, Z.; Wan, Y.; Yao, Y. miRNA-21 promotes proliferation and invasion of triple-negative breast cancer cells through targeting PTEN. Am. J. Transl. Res. 2017, 9, 953–961. [Google Scholar]
- Hong, H.C.; Chuang, C.H.; Huang, W.C.; Weng, S.L.; Chen, C.H.; Chang, K.H.; Liao, K.W.; Huang, H.D. A panel of eight microRNAs is a good predictive parameter for triple-negative breast cancer relapse. Theranostics 2020, 10, 8771–8789. [Google Scholar] [CrossRef]
- Ren, Y.Q.; Fu, F.; Han, J. MiR-27a modulates radiosensitivity of triple-negative breast cancer (TNBC) cells by targeting CDC27. Med. Sci. Monit. 2015, 21, 1297–1303. [Google Scholar]
- Masoudi-Khoram, N.; Abdolmaleki, P.; Hosseinkhan, N.; Nikoofar, A.; Mowla, S.J.; Monfared, H.; Baldassarre, G. Differential miRNAs expression pattern of irradiated breast cancer cell lines is correlated with radiation sensitivity. Sci. Rep. 2020, 10, 9054. [Google Scholar] [CrossRef]
- To, N.H.; Nguyen, H.Q.; Thiolat, A.; Liu, B.; Cohen, J.; Radosevic-Robin, N.; Belkacemi, Y.; TransAtlantic Radiation Oncology Network (TRONE); Association of Radiotherapy and Oncology of the Mediterranean Area (AROME). Radiation therapy for triple-negative breast cancer: Emerging role of microRNAs as biomarkers and radiosensitivity modifiers. A systematic review. Breast Cancer Res. Treat. 2022, 193, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Lan, J.; Luo, H.; Fu, H.; Xiao, X.; Yang, L. FOS-Mediated PLCB1 Induces Radioresistance and Weakens the Antitumor Effects of CD8(+) T Cells in Triple-Negative Breast Cancer. Mol. Carcinog. 2025, 64, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Musielak, M.; Graczyk, K.; Liszka, M.; Christou, A.; Rosochowicz, M.A.; Lach, M.S.; Adamczyk, B.; Suchorska, W.M.; Piotrowski, T.; Stenerlöw, B.; et al. Impact of Proton Irradiation Depending on Breast Cancer Subtype in Patient-Derived Cell Lines. Int. J. Mol. Sci. 2024, 25, 10494. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Griffith, J.; Panneerselvam, J.; Babu, A.; Mani, J.; Herman, T.; Ramesh, R.; Munshi, A. Regorafenib sensitizes human breast cancer cells to radiation by inhibiting multiple kinases and inducing DNA damage. Int. J. Radiat. Biol. 2021, 97, 1109–1120. [Google Scholar] [CrossRef]
- Choi, E.; Jeon, K.H.; Lee, H.; Mun, G.I.; Kim, J.A.; Shin, J.H.; Kwon, Y.; Na, Y.; Lee, Y.S. Radiosensitizing effect of a novel CTSS inhibitor by enhancing BRCA1 protein stability in triple-negative breast cancer cells. Cancer Sci. 2024, 115, 2036–2048. [Google Scholar] [CrossRef]
- Liu, Z.; Li, M.; Jiang, Z.; Wang, X. A Comprehensive Immunologic Portrait of Triple-Negative Breast Cancer. Transl. Oncol. 2018, 11, 311–329. [Google Scholar] [CrossRef]
- Demaria, S.; Bhardwaj, N.; McBride, W.H.; Formenti, S.C. Combining radiotherapy and immunotherapy: A revived partnership. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 655–666. [Google Scholar] [CrossRef]
- Gong, J.; Le, T.Q.; Massarelli, E.; Hendifar, A.E.; Tuli, R. Radiation therapy and PD-1/PD-L1 blockade: The clinical development of an evolving anticancer combination. J. Immunother. Cancer 2018, 6, 46. [Google Scholar] [CrossRef]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Ren, Y.; Song, J.; Li, X.; Luo, N. Rationale and Clinical Research Progress on PD-1/PD-L1-Based Immunotherapy for Metastatic Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 8878. [Google Scholar] [CrossRef]
- Hu, Z.I.; Ho, A.Y.; McArthur, H.L. Combined Radiation Therapy and Immune Checkpoint Blockade Therapy for Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 153–164. [Google Scholar] [CrossRef]
- Azoury, F.; Misra, S.; Barry, A.; Helou, J. Role of radiation therapy in triple negative breast cancer: Current state and future directions—A narrative review. Precis. Cancer Med. 2021, 5, 3. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune Cells | Role in TNBC TME | Ref. |
|---|---|---|
| TILs | High infiltration of TILs is observed in TNBC-TME and is associated with neoadjuvant chemotherapy (NAC) response. | [44] |
| TILs | Stromal lymphocytic infiltration increased prognostic value in TNBC patients. | [51] |
| TILs | Infiltration of B cells and B cell marker expression predominantly associated with predicting prognosis and response to immunotherapy in TNBC patients. | [49] |
| TILs | T-cell infiltration and a high ratio of CD4/FOXP3 and CD8/CD163 proteins improve one-year overall survival in metastatic TNBC patients. | [50] |
| TILs | Increased CD8+T cell infiltration in TNBC xenograft tumors suppresses PD-L1 expression when mice fed vitamin C and LIVI overexpression modulate this process. | [46] |
| MDSCs | Inhibition of MDSCs biosynthesis enhances immunotherapy efficacy by myeloid maturation and activation (CD8+ T cell) in BC. | [60] |
| MDSCs | Immunosuppressive MDSCs accumulation in the TNBC 4T1 cell tumor mouse model was observed and NKT cell activation via DCs decreased MDSCs immunosuppressive activity. | [61] |
| Macrophages | Higher levels of TAMs are found in TNBC TME and macrophage colony-stimulating factors (M-CSF) and IL-6 drive macrophages toward M2 polarization and infiltration. | [67,69,70] |
| Macrophages | Higher numbers of M2 CD163+ and CD68+ macrophages are present in TNBC/basal-like breast cancer compared to luminal types. | [70] |
| Macrophages | Infiltration of higher densities of CD163+ macrophage TNBC tumors improved OS and BC-specific survival independently in invasive TNBC. | [75] |
| Macrophages | By activation of CCL2/AKT/β-catenin signaling, TAM-M2 stimulates EMT and cancer stem cell (CSC) properties in TNBC. | [76] |
| CAFs | The presence of CAFs in TNBC TME promotes TNBC progression by the activation of TGF-β. | [81] |
| CAFs | Myeloid cells mediated expression of CXCL16 activates CAFs and promotes fibroblast infiltration in TNBC TME. | [83] |
| CAFs | High expression of CAF-related G protein-coupled receptor 34 (CAF-GPR34) in TNBC patients serves as a prognosis biomarker in response to immunotherapy. | [84] |
| TANs | TNBC tumor cells release GM-CSF, TGF-β, and CXCR2 stimulate TANs to release tumor suppressor M, promote angiogenesis, and improve tumor cell infiltration or recruitment of neutrophils in TNBC-TME. | [87,88,89] |
| NK cells | By downregulation MHC-I, NK cells recognize TNBC tumor cells and ICIs, and cytokine stimulation restores NK cell activity in TNBC-TME. | [91,95,96] |
| NK cells | CAR-NK cells targeting HER1, engineered with catalase in TNBC-TME, modulate cytotoxic potential and prevent postoperative local and distant relapses of TNBC tumors. | [97] |
| CAAs | By the induction of CCL5, CAAs increased the invasiveness of TNBC MDA-MB-231 cells. | [103] |
| CAAs | Exposure of hesperidin to CAA inhibits CCL2, elevates ADPN secretion, reduces recruitment of M2 macrophages, and potentiates the efficacy of PD-1 in TNBC TME. | [106] |
| CAAs | In TNBC-TME, CAA meditated secretion of CXCL8 suppressed CD4+ T and CD8+ T and upregulating CD274 suggests targeting the CXCL8 and PD-1 inhibition synergistically increased the tumor immune response. | [107] |
| Agents | Target | Mechanisms | Ref. |
|---|---|---|---|
| RT | TILs, TAMs | Higher infiltration in TNBC TME | [125] |
| RT | CXCR6, CD8+ T cells | Elevated CXCR6 regulates CD8+T differentiation leads to superior response to adjuvant radiotherapy and immunotherapy in TNBC. | [137] |
| RT | EGFR | Migration of CAR-T cells in TME, activates the NF-κB, and induces ICAM1 that regulates antitumor effects in TNBC | [139] |
| Hypofrac. RT (HFRT) | MDSCs | Combined HFRT, and CXCR2 blockade inhibits MDSCs infiltration in TNBC tumors and increases the efficacy of CAR-T cells | [147] |
| Radiolabeled biomolecule(RB) + RT + anti-PDL1 and-CTLA4 (CP) | Macrophages MDSCs | RB + RT + CP suppressed macrophages and MDSCs, infiltration in TNBC tumors that contributes to immune escape and tumor relapse. | [148] |
| TAU+ RT | CD151 (T cell activator) | Transcriptional downregulation of CD151 | [149] |
| RT | NK cells | NK cell migration and penetration into the primary TNBC tumor reduced tumor burden and growth | [140] |
| PI3Kαδ Inhibition. +RT | PI3Kαδ and PI3Kγδ | Reduce TNBC tumor hypoxia and Antitumor Immune Effect of Anti-PD1 and RT sensitization | [141] |
| PI3KδγInhibition +RT | CD8+T | Potentiates effector CD8(+) T cell-dependent antitumor and abscopal effect after RT | [142] |
| RT + anti-PD-1 | PD-1 | Reduced TNBC tumor growth and metastasis in mice | [143] |
| RT + siRNA | PD-L1 | TNBC 4T1 derived cell membrane (CM) coated PD-L1 siRNA-decorated Au/MnO2 nanosensitizer (R&F@Au/MnO2-CM) enhances radio-immunotherapy synergistically. | [150] |
| RT + anti CTLA-4 | CTLA-4 | Inhibit TNBC lung metastasis and increase survival in mice | [144] |
| Olaparib +RT | PARP | Improve clinical responses | [151,152] |
| Pembrolizumab +RT | PD-1 | Immunosuppressive, and safe for mTNBC PD-L1 negative patients | [153] |
| TAlazoparib + RT | PARP | Increased tumor PD-L1 expression enhanced sensitivity to PD-L1 inhibitor, atezolizumab. | [154] |
| Agents | Target | Mechanisms | Ref. |
|---|---|---|---|
| RT | Elongin B | Reduced mitochondrial oxygen consumption rate. | [164] |
| RT | XRCC4 | Knockdown increased radiation-mediated DNA damage. | [165] |
| CFI-400945 + RT | Polo-like Kinase 4 (PLK4) | Increase antiproliferative/radio sensitization by overamplification of centrioles. | [166] |
| RT | BUB1 (cell cycle Ser/Thr kinase) | BUB1 deletion impaired RT-mediated DSBs repair by recruitment of phospho-, total-DNAPK, and KAP1 to chromatin. | [168] |
| RT | Threonine tyrosine kinase inhibition (TTKi) | TTK knockdown or inhibition reduces tumor growth in vivo. | [169] |
| RT | RAD50, a DNA repair protein | Silencing of RAD50 by siRNA nanoparticles, RT enhances cell apoptosis | [170] |
| ZR-BA1+ RT | EGFR | Induced DSBs and impaired DNA repair | [171] |
| OTSSP167 + RT | MELK | Inactivation/deletion of MELK increased radiation sensitivity and inhibited tumor growth | [172] |
| RT | miR-27 | MiR-27 targets CDC27 and involves the radiosensitivity of TNBC cells | [179] |
| RT | FOS/Phospholipase C beta 1 (PLCB1) | RT mediates FOS/PLCB1-induced radioresistance by impairing CD8+ T cell activity and by the activation of PI3K/AKT signaling pathway. | [182] |
| RT | γH2AX and p53 | Increased DNA damage in MCF-7 BC compared to the TNBC and CAFs extracted from the tumor tissue (TNBC subtype tumor) shows increased resistance to ionizing compared to luminal A tumors isolated. | [183] |
| RT + Regorafenib | Multi-kinase inhibitor | Regorafenib enhanced the radiosensitivity of TNBC but not MCF 10a normal breast cells. | [184] |
| RT | Cathepsin S (CTSS) | Inhibition of CTSS restores BRCA1 function and enhances RT-induced apoptosis of TNBC cells. | [185] |
| RT | miR-16-5p and miR-23b-3p | RT induces miR-16-5p in breast cancer but not in TNBC and is associated with radiation response | [180] |
| RT | miR-21, miR-33a, miR-139-5p, and miR-210 | Associated with radiation response in TNBC patients | [181] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niture, S.; Ghosh, S.; Jaboin, J.; Seneviratne, D. Tumor Microenvironment Dynamics of Triple-Negative Breast Cancer Under Radiation Therapy. Int. J. Mol. Sci. 2025, 26, 2795. https://doi.org/10.3390/ijms26062795
Niture S, Ghosh S, Jaboin J, Seneviratne D. Tumor Microenvironment Dynamics of Triple-Negative Breast Cancer Under Radiation Therapy. International Journal of Molecular Sciences. 2025; 26(6):2795. https://doi.org/10.3390/ijms26062795
Chicago/Turabian StyleNiture, Suryakant, Subhajit Ghosh, Jerry Jaboin, and Danushka Seneviratne. 2025. "Tumor Microenvironment Dynamics of Triple-Negative Breast Cancer Under Radiation Therapy" International Journal of Molecular Sciences 26, no. 6: 2795. https://doi.org/10.3390/ijms26062795
APA StyleNiture, S., Ghosh, S., Jaboin, J., & Seneviratne, D. (2025). Tumor Microenvironment Dynamics of Triple-Negative Breast Cancer Under Radiation Therapy. International Journal of Molecular Sciences, 26(6), 2795. https://doi.org/10.3390/ijms26062795