
Fine Regulation of MicroRNAs in Gene Regulatory Networks and Pathophysiology


Abstract
1. Introduction
2. miRNA Biogenesis
2.1. Canonical miRNA Biogenesis
2.2. Mutations in miRNA Biogenesis Pathway and Associated Human Diseases
2.2.1. DGCR8 (DiGeorge Syndrome Critical Region 8)
2.2.2. AGO (Argonaute)
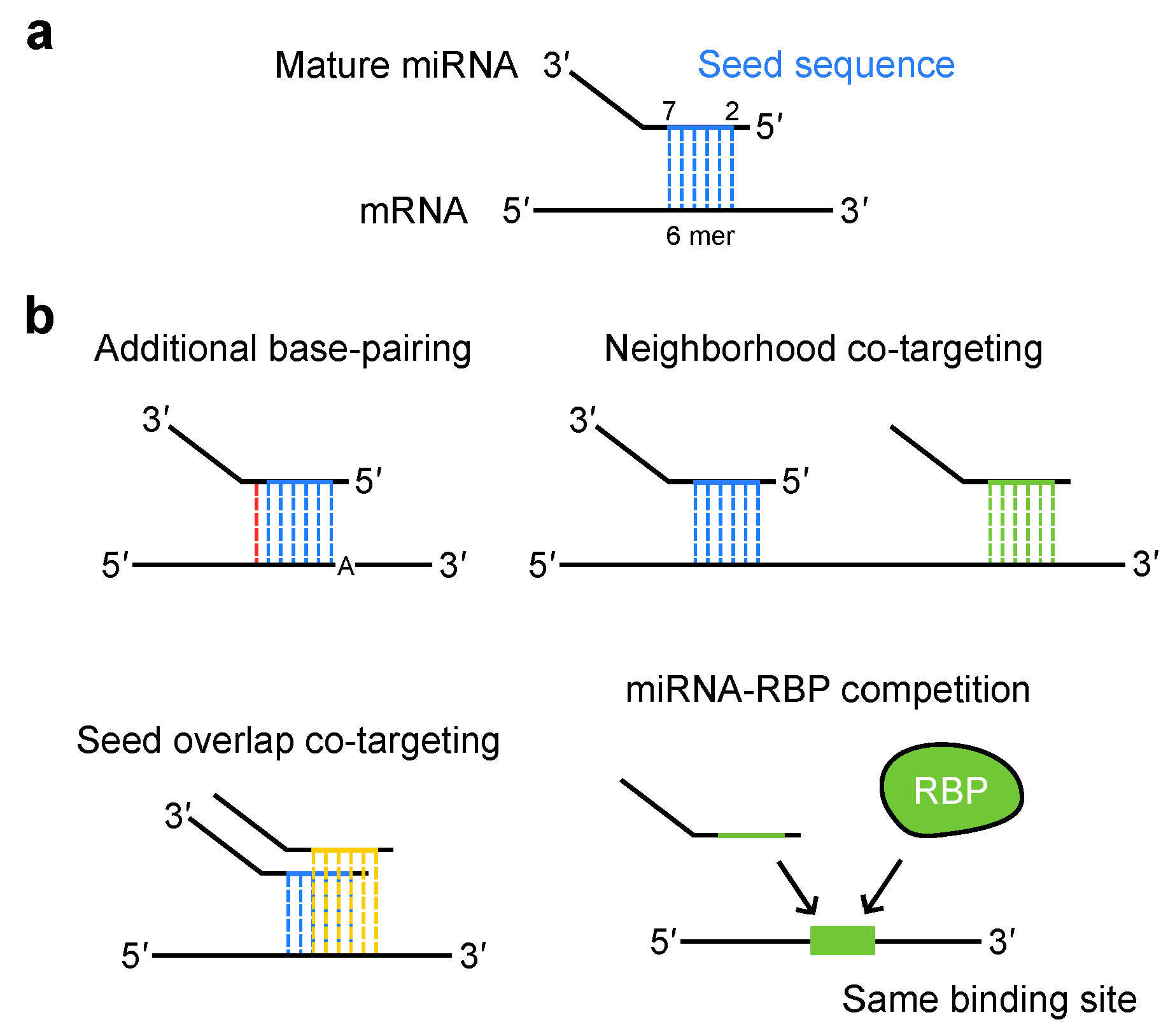
3. Diverse Modes of miRNA Targeting
3.1. miRNA Target Recognition
3.2. miRNA Co-Targeting
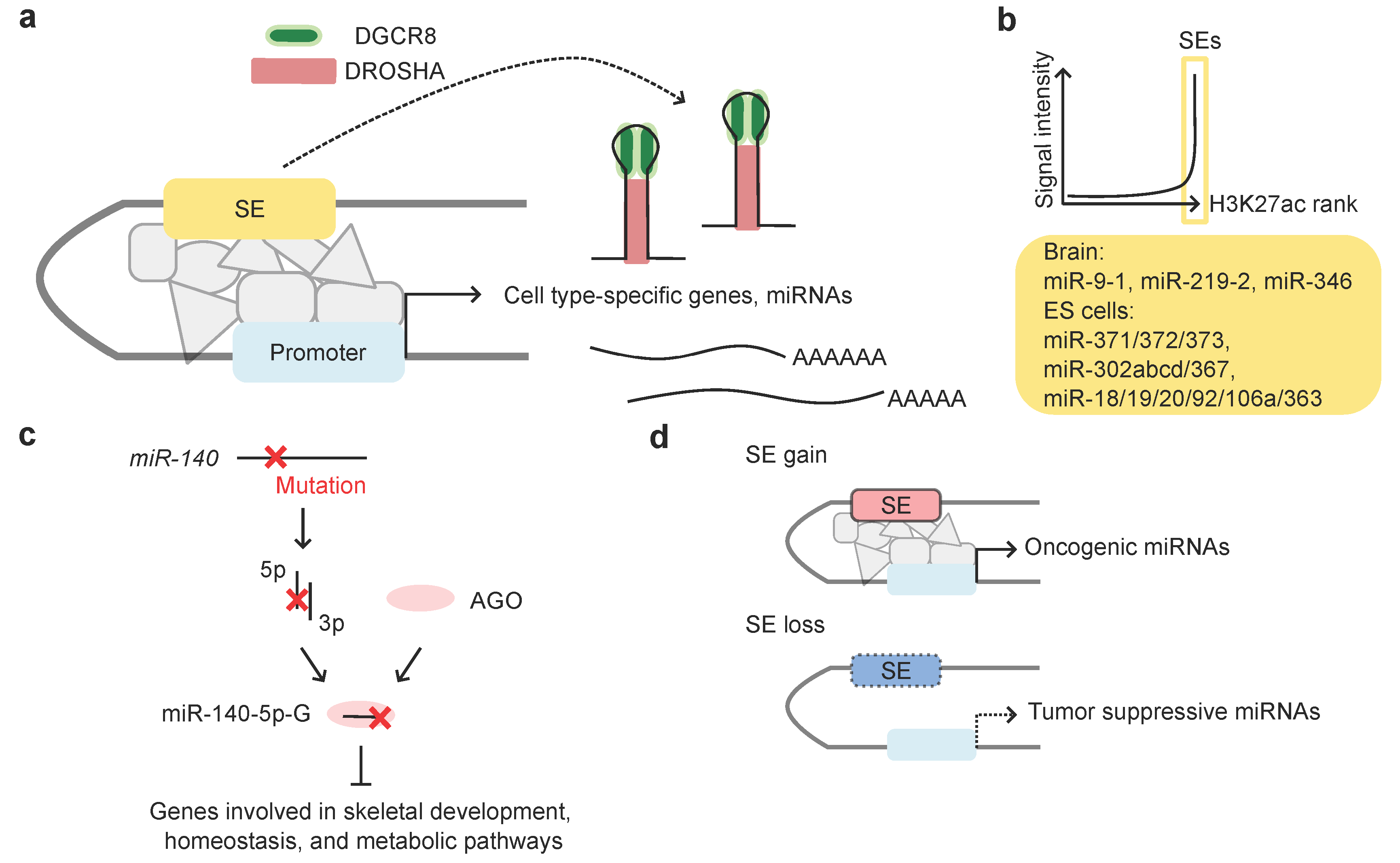
4. Super-Enhancers (SEs) Drive Cell Type-Specific miRNA Expression
4.1. Super-Enhancers (SEs) and miRNAs
4.2. SE-Associated miRNAs and Human Diseases
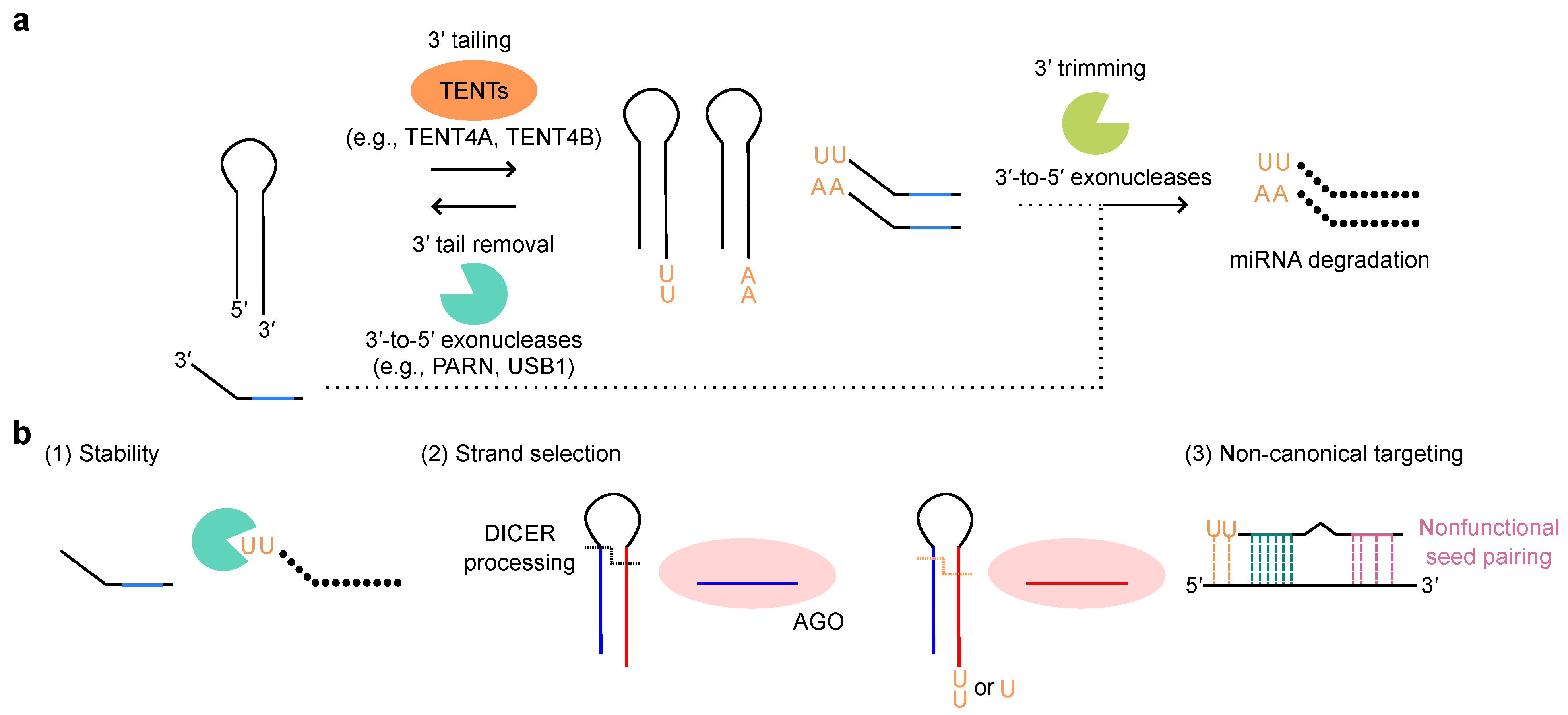
5. The Post-Transcription Regulation of miRNA Expression: 3′ Tailing, 3′ Trimming, and TDMD
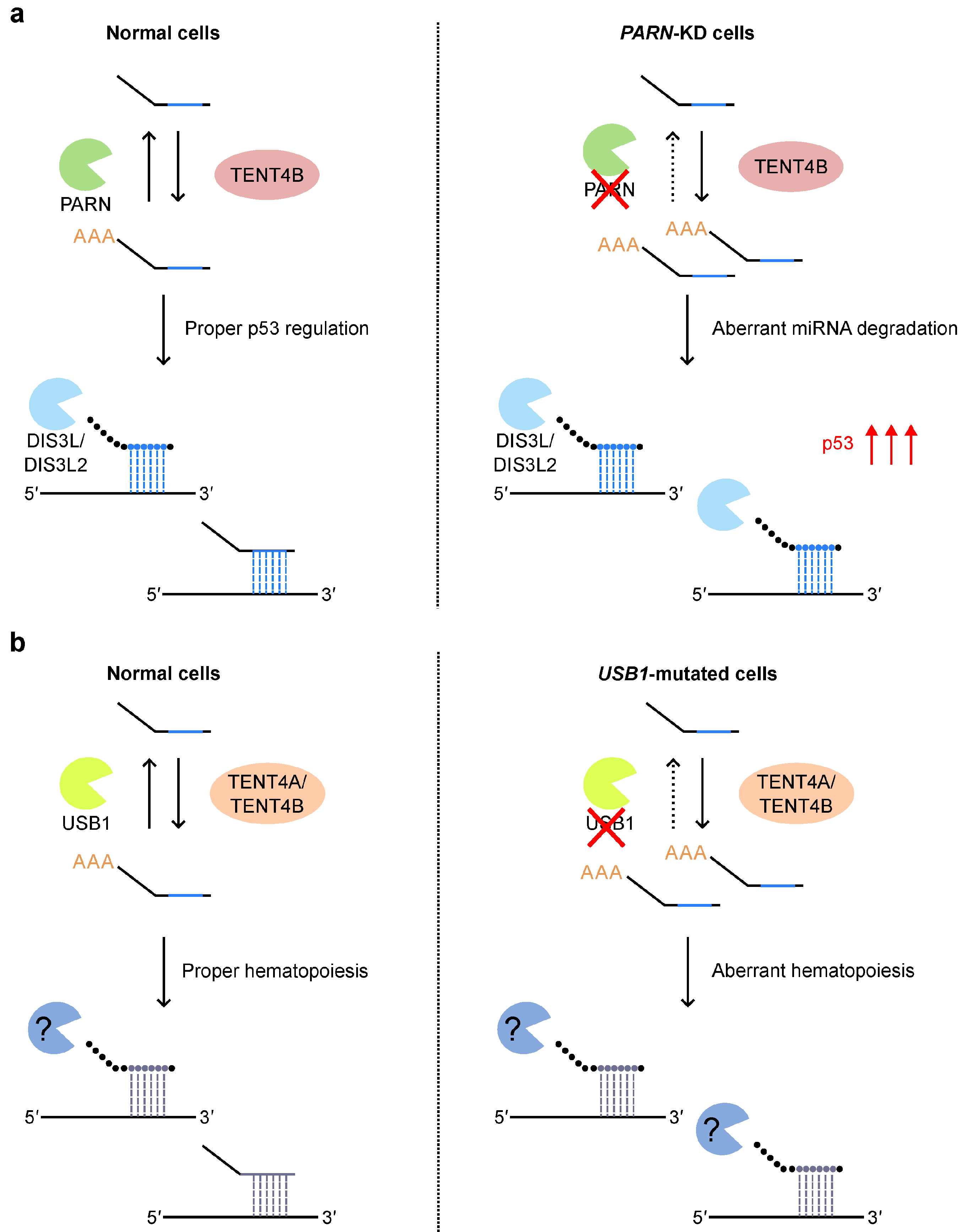
5.1. miRNA 3′ Tailing and 3′ Trimming: Post-Transcriptional Modifications of miRNAs
5.2. Target-Directed miRNA Degradation (TDMD)
5.3. miRNA 3′ Tailing and 3′ Trimming in Human Diseases
6. The Evolution of miRNA Genes and Its Potential Impacts
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| UTR | Untranslated region |
| SE | Super-enhancer |
| TF | Transcription factor |
| TDMD | Target-directed miRNA degradation |
| pri-miRNA | Primary miRNA |
| pre-miRNA | Precursor miRNA |
| XPO5 | Exportin-5 |
| AGO | Argonaute |
| RISC | RNA-induced silencing complex |
| NDD | Neurological development disorder |
| ID | Intellectual disability |
| ASD | Autism spectrum disorder |
| DC | Dyskeratosis congenita |
| PN | Poikiloderma with neutropenia |
| RNA-seq | RNA sequencing |
| SLE | Systemic lupus erythematosus |
| RBP | RNA-binding protein |
| ESC | Embryonic stem cell |
| GOF | Gain-of-function |
| ChIP-seq | Chromatin immunoprecipitation-sequencing |
| TCGA | The Cancer Genome Atlas |
| SCLC | Small cell lung cancer |
| TENT | Terminal nucleotidyl transferase |
| TE | Typical enhancer |
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene Lin-14 by Lin-4 Mediates Temporal Pattern Formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Horvitz, H.R. Heterochronic Mutants of the Nematode Caenorhabditis elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef]
- Ambros, V. A Hierarchy of Regulatory Genes Controls a Larva-to-Adult Developmental Switch in C. elegans. Cell 1989, 57, 49–57. [Google Scholar] [CrossRef]
- Wightman, B.; Bürglin, T.R.; Gatto, J.; Arasu, P.; Ruvkun, G. Negative Regulatory Sequences in the Lin-14 3′-Untranslated Region Are Necessary to Generate a Temporal Switch during Caenorhabditis elegans Development. Genes Dev. 1991, 5, 1813–1824. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-Nucleotide Let-7 RNA Regulates Developmental Timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the Sequence and Temporal Expression of Let-7 Heterochronic Regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef]
- Lee, R.C.; Ambros, V. An Extensive Class of Small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.; Martienssen, R.A. The Expanding World of Small RNAs in Plants. Nat. Rev. Mol. Cell Biol. 2015, 16, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Strzyz, P. microRNA Communication in Plants. Nat. Rev. Mol. Cell Biol. 2021, 22, 775. [Google Scholar] [CrossRef] [PubMed]
- Ninkuu, V.; Liu, Z.; Sun, X. Genetic Regulation of Nitrogen Use Efficiency in Gossypium spp. Plant Cell Environ. 2023, 46, 1749–1773. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chiou, T.-J.; Lin, S.-I.; Aung, K.; Zhu, J.-K. A miRNA Involved in Phosphate-Starvation Response in Arabidopsis. Curr. Biol. 2005, 15, 2038–2043. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Young, R.A.; Sharp, P.A. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 2017, 168, 1000–1014.e15. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Yu, S.; Kim, V.N. A Tale of Non-Canonical Tails: Gene Regulation by Post-Transcriptional RNA Tailing. Nat. Rev. Mol. Cell Biol. 2020, 21, 542–556. [Google Scholar] [CrossRef]
- Komatsu, S.; Kitai, H.; Suzuki, H.I. Network Regulation of microRNA Biogenesis and Target Interaction. Cells 2023, 12, 306. [Google Scholar] [CrossRef]
- Suzuki, H.I. Roles of MicroRNAs in Disease Biology. JMA J. 2023, 6, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA Genes Are Transcribed by RNA Polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Baek, S.C.; Lee, Y.-Y.; Bastiaanssen, C.; Kim, J.; Kim, H.; Kim, V.N. A Quantitative Map of Human Primary microRNA Processing Sites. Mol. Cell 2021, 81, 3422–3439.e11. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The Nuclear RNase III Drosha Initiates microRNA Processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor Complex Mediates the Genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of Primary microRNAs by the Microprocessor Complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 Mediates the Nuclear Export of Pre-microRNAs and Short Hairpin RNAs. Genes. Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and Mechanisms Related to RNA Interference Regulate Expression of the Small Temporal RNAs That Control C. elegans Developmental Timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Ketting, R.F.; Fischer, S.E.J.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H.A. Dicer Functions in RNA Interference and in Synthesis of Small RNA Involved in Developmental Timing in C. elegans. Genes. Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef]
- Knight, S.W.; Bass, B.L. A Role for the RNase III Enzyme DCR-1 in RNA Interference and Germ Line Development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, É.; Tuschl, T.; Zamore, P.D. A Cellular Function for the RNA-Interference Enzyme Dicer in the Maturation of the Let-7 Small Temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kolb, F.A.; Jaskiewicz, L.; Westhof, E.; Filipowicz, W. Single Processing Center Models for Human Dicer and Bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, P.J.F.; Ameres, S.L.; Kueng, S.; Martinez, J. Cleavage of the siRNA Passenger Strand during RISC Assembly in Human Cells. EMBO Rep. 2006, 7, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs Exhibit Strand Bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Katsura, A.; Yasuda, T.; Ueno, T.; Mano, H.; Sugimoto, K.; Miyazono, K. Small-RNA Asymmetry Is Directly Driven by Mammalian Argonautes. Nat. Struct. Mol. Biol. 2015, 22, 512–521. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-Directed Nuclease Mediates Post-Transcriptional Gene Silencing in Drosophila Cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a Link Between Genetic and Biochemical Analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef]
- Mourelatos, Z.; Dostie, J.; Paushkin, S.; Sharma, A.; Charroux, B.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. miRNPs: A Novel Class of Ribonucleoproteins Containing Numerous microRNAs. Genes. Dev. 2002, 16, 720–728. [Google Scholar] [CrossRef]
- Pelletier, D.; Rivera, B.; Fabian, M.R.; Foulkes, W.D. miRNA Biogenesis and Inherited Disorders: Clinico-Molecular Insights. Trends Genet. 2023, 39, 401–414. [Google Scholar] [CrossRef]
- Rakheja, D.; Chen, K.S.; Liu, Y.; Shukla, A.A.; Schmid, V.; Chang, T.-C.; Khokhar, S.; Wickiser, J.E.; Karandikar, N.J.; Malter, J.S.; et al. Somatic Mutations in DROSHA and DICER1 Impair microRNA Biogenesis through Distinct Mechanisms in Wilms Tumours. Nat. Commun. 2014, 5, 4802. [Google Scholar] [CrossRef]
- Torrezan, G.T.; Ferreira, E.N.; Nakahata, A.M.; Barros, B.D.F.; Castro, M.T.M.; Correa, B.R.; Krepischi, A.C.V.; Olivieri, E.H.R.; Cunha, I.W.; Tabori, U.; et al. Recurrent Somatic Mutation in DROSHA Induces microRNA Profile Changes in Wilms Tumour. Nat. Commun. 2014, 5, 4039. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.L.; Ooms, A.; Gadd, S.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.-R.; Hsu, C.H.; Yan, C.; et al. Recurrent DGCR8, DROSHA, and SIX Homeodomain Mutations in Favorable Histology Wilms Tumors. Cancer Cell 2015, 27, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Wegert, J.; Ishaque, N.; Vardapour, R.; Geörg, C.; Gu, Z.; Bieg, M.; Ziegler, B.; Bausenwein, S.; Nourkami, N.; Ludwig, N.; et al. Mutations in the SIX1/2 Pathway and the DROSHA/DGCR8 miRNA Microprocessor Complex Underlie High-Risk Blastemal Type Wilms Tumors. Cancer Cell 2015, 27, 298–311. [Google Scholar] [CrossRef] [PubMed]
- de la Chapelle, A.; Herva, R.; Koivisto, M.; Aula, P. A Deletion in Chromosome 22 Can Cause DiGeorge Syndrome. Hum. Genet. 1981, 57, 253–256. [Google Scholar] [CrossRef]
- Kelley, R.I.; Zackai, E.H.; Emanuel, B.S.; Kistenmacher, M.; Greenberg, F.; Punnett, H.H. The Association of the DiGeorge Anomalad with Partial Monosomy of Chromosome 22. J. Pediatr. 1982, 101, 197–200. [Google Scholar] [CrossRef]
- Driscoll, D.A.; Spinner, N.B.; Budarf, M.L.; McDonald-McGinn, D.M.; Zackai, E.H.; Goldberg, R.B.; Shprintzen, R.J.; Saal, H.M.; Zonana, J.; Jones, M.C. Deletions and Microdeletions of 22q11.2 in Velo-Cardio-Facial Syndrome. Am. J. Med. Genet. 1992, 44, 261–268. [Google Scholar] [CrossRef]
- Driscoll, D.A.; Budarf, M.L.; Emanuel, B.S. A Genetic Etiology for DiGeorge Syndrome: Consistent Deletions and Microdeletions of 22q11. Am. J. Hum. Genet. 1992, 50, 924–933. [Google Scholar]
- Karayiorgou, M.; Morris, M.A.; Morrow, B.; Shprintzen, R.J.; Goldberg, R.; Borrow, J.; Gos, A.; Nestadt, G.; Wolyniec, P.S.; Lasseter, V.K. Schizophrenia Susceptibility Associated with Interstitial Deletions of Chromosome 22q11. Proc. Natl. Acad. Sci. USA 1995, 92, 7612–7616. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Primers 2015, 1, 1–19. [Google Scholar] [CrossRef]
- Rivera, B.; Nadaf, J.; Fahiminiya, S.; Apellaniz-Ruiz, M.; Saskin, A.; Chong, A.-S.; Sharma, S.; Wagener, R.; Revil, T.; Condello, V.; et al. DGCR8 Microprocessor Defect Characterizes Familial Multinodular Goiter with Schwannomatosis. J. Clin. Investig. 2020, 130, 1479–1490. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and Mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 Mutations in Familial Pleuropulmonary Blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef]
- Heravi-Moussavi, A.; Anglesio, M.S.; Cheng, S.-W.G.; Senz, J.; Yang, W.; Prentice, L.; Fejes, A.P.; Chow, C.; Tone, A.; Kalloger, S.E.; et al. Recurrent Somatic DICER1 Mutations in Nonepithelial Ovarian Cancers. N. Engl. J. Med. 2012, 366, 234–242. [Google Scholar] [CrossRef]
- González, I.A.; Stewart, D.R.; Schultz, K.A.P.; Field, A.P.; Hill, D.A.; Dehner, L.P. DICER1 Tumor Predisposition Syndrome: An Evolving Story Initiated with the Pleuropulmonary Blastoma. Mod. Pathol. 2022, 35, 4–22. [Google Scholar] [CrossRef]
- Kommoss, F.K.F.; Chong, A.-S.; Chong, A.-L.; Pfaff, E.; Jones, D.T.W.; Hiemcke-Jiwa, L.S.; Kester, L.A.; Flucke, U.; Gessler, M.; Schrimpf, D.; et al. Genomic Characterization of DICER1-Associated Neoplasms Uncovers Molecular Classes. Nat. Commun. 2023, 14, 1677. [Google Scholar] [CrossRef]
- Wu, M.; Sabbaghian, N.; Xu, B.; Addidou-Kalucki, S.; Bernard, C.; Zou, D.; Reeve, A.; Eccles, M.; Cole, C.; Choong, C.; et al. Biallelic DICER1 Mutations Occur in Wilms Tumours. J. Pathol. 2013, 230, 154–164. [Google Scholar] [CrossRef]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of Genetic Mutations Associated with Severe Non-Syndromic Sporadic Intellectual Disability: An Exome Sequencing Study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De Novo Mutations in Moderate or Severe Intellectual Disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef]
- Tokita, M.J.; Chow, P.M.; Mirzaa, G.; Dikow, N.; Maas, B.; Isidor, B.; Le Caignec, C.; Penney, L.S.; Mazzotta, G.; Bernardini, L.; et al. Five Children with Deletions of 1p34.3 Encompassing AGO1 and AGO3. Eur. J. Hum. Genet. 2015, 23, 761–765. [Google Scholar] [CrossRef]
- Martínez, F.; Caro-Llopis, A.; Roselló, M.; Oltra, S.; Mayo, S.; Monfort, S.; Orellana, C. High Diagnostic Yield of Syndromic Intellectual Disability by Targeted Next-Generation Sequencing. J. Med. Genet. 2017, 54, 87–92. [Google Scholar] [CrossRef]
- Sakaguchi, A.; Yamashita, Y.; Ishii, T.; Uehara, T.; Kosaki, K.; Takahashi, T.; Takenouchi, T. Further Evidence of a Causal Association between AGO1, a Critical Regulator of microRNA Formation, and Intellectual Disability/Autism Spectrum Disorder. Eur. J. Med. Genet. 2019, 62, 103537. [Google Scholar] [CrossRef] [PubMed]
- Lessel, D.; Zeitler, D.M.; Reijnders, M.R.F.; Kazantsev, A.; Hassani Nia, F.; Bartholomäus, A.; Martens, V.; Bruckmann, A.; Graus, V.; McConkie-Rosell, A.; et al. Germline AGO2 Mutations Impair RNA Interference and Human Neurological Development. Nat. Commun. 2020, 11, 5797. [Google Scholar] [CrossRef]
- Schalk, A.; Cousin, M.A.; Dsouza, N.R.; Challman, T.D.; Wain, K.E.; Powis, Z.; Minks, K.; Trimouille, A.; Lasseaux, E.; Lacombe, D.; et al. De Novo Coding Variants in the AGO1 Gene Cause a Neurodevelopmental Disorder with Intellectual Disability. J. Med. Genet. 2022, 59, 965–975. [Google Scholar] [CrossRef]
- Tummala, H.; Walne, A.; Collopy, L.; Cardoso, S.; de la Fuente, J.; Lawson, S.; Powell, J.; Cooper, N.; Foster, A.; Mohammed, S.; et al. Poly(A)-Specific Ribonuclease Deficiency Impacts Telomere Biology and Causes Dyskeratosis Congenita. J. Clin. Investig. 2015, 125, 2151–2160. [Google Scholar] [CrossRef]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome Sequencing Links Mutations in PARN and RTEL1 with Familial Pulmonary Fibrosis and Telomere Shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef]
- Burris, A.M.; Ballew, B.J.; Kentosh, J.B.; Turner, C.E.; Norton, S.A.; Bass, S.; Boland, J.; Burdett, L.; Chowdhury, S.; Cullen, M.; et al. Hoyeraal-Hreidarsson Syndrome Due to PARN Mutations: Fourteen Years of Follow-Up. Pediatr. Neurol. 2016, 56, 62–68.e1. [Google Scholar] [CrossRef]
- Shukla, S.; Bjerke, G.A.; Muhlrad, D.; Yi, R.; Parker, R. The RNase PARN Controls the Levels of Specific miRNAs That Contribute to P53 Regulation. Mol. Cell 2019, 73, 1204–1216.e4. [Google Scholar] [CrossRef]
- Huynh, T.N.; Parker, R. The PARN, TOE1, and USB1 RNA Deadenylases and Their Roles in Non-Coding RNA Regulation. J. Biol. Chem. 2023, 299, 105139. [Google Scholar] [CrossRef]
- Volpi, L.; Roversi, G.; Colombo, E.A.; Leijsten, N.; Concolino, D.; Calabria, A.; Mencarelli, M.A.; Fimiani, M.; Macciardi, F.; Pfundt, R.; et al. Targeted Next-Generation Sequencing Appoints C16orf57 as Clericuzio-Type Poikiloderma with Neutropenia Gene. Am. J. Hum. Genet. 2010, 86, 72–76. [Google Scholar] [CrossRef]
- Colombo, E.A.; Bazan, J.F.; Negri, G.; Gervasini, C.; Elcioglu, N.H.; Yucelten, D.; Altunay, I.; Cetincelik, U.; Teti, A.; Del Fattore, A.; et al. Novel C16orf57 Mutations in Patients with Poikiloderma with Neutropenia: Bioinformatic Analysis of the Protein and Predicted Effects of All Reported Mutations. Orphanet J. Rare Dis. 2012, 7, 7. [Google Scholar] [CrossRef]
- Jeong, H.-C.; Shukla, S.; Fok, W.C.; Huynh, T.N.; Batista, L.F.Z.; Parker, R. USB1 Is a miRNA Deadenylase That Regulates Hematopoietic Development. Science 2023, 379, 901–907. [Google Scholar] [CrossRef]
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the Contemporary Live-Birth Prevalence of Recurrent 22q11.2 Deletions: A Cross-Sectional Analysis from Population-Based Newborn Screening. Can. Med. Assoc. Open Access J. 2021, 9, E802–E809. [Google Scholar] [CrossRef]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered Brain microRNA Biogenesis Contributes to Phenotypic Deficits in a 22q11-Deletion Mouse Model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Mukai, J.; Dhilla, A.; Drew, L.J.; Stark, K.L.; Cao, L.; MacDermott, A.B.; Karayiorgou, M.; Gogos, J.A. Palmitoylation-Dependent Neurodevelopmental Deficits in a Mouse Model of 22q11 Microdeletion. Nat. Neurosci. 2008, 11, 1302–1310. [Google Scholar] [CrossRef]
- Fénelon, K.; Mukai, J.; Xu, B.; Hsu, P.-K.; Drew, L.J.; Karayiorgou, M.; Fischbach, G.D.; MacDermott, A.B.; Gogos, J.A. Deficiency of Dgcr8, a Gene Disrupted by the 22q11.2 Microdeletion, Results in Altered Short-Term Plasticity in the Prefrontal Cortex. Proc. Natl. Acad. Sci. USA 2011, 108, 4447–4452. [Google Scholar] [CrossRef]
- Earls, L.R.; Fricke, R.G.; Yu, J.; Berry, R.B.; Baldwin, L.T.; Zakharenko, S.S. Age-Dependent MicroRNA Control of Synaptic Plasticity in 22q11 Deletion Syndrome and Schizophrenia. J. Neurosci. 2012, 32, 14132–14144. [Google Scholar] [CrossRef]
- Toyoshima, M.; Akamatsu, W.; Okada, Y.; Ohnishi, T.; Balan, S.; Hisano, Y.; Iwayama, Y.; Toyota, T.; Matsumoto, T.; Itasaka, N.; et al. Analysis of Induced Pluripotent Stem Cells Carrying 22q11.2 Deletion. Transl. Psychiatry 2016, 6, e934. [Google Scholar] [CrossRef]
- Chun, S.; Du, F.; Westmoreland, J.J.; Han, S.B.; Wang, Y.-D.; Eddins, D.; Bayazitov, I.T.; Devaraju, P.; Yu, J.; Mellado Lagarde, M.M.; et al. Thalamic miR-338-3p Mediates Auditory Thalamocortical Disruption and Its Late Onset in Models of 22q11.2 Microdeletion. Nat. Med. 2017, 23, 39–48. [Google Scholar] [CrossRef]
- Ying, S.; Heung, T.; Zhang, Z.; Yuen, R.K.C.; Bassett, A.S. Schizophrenia Risk Mediated by microRNA Target Genes Overlapped by Genome-Wide Rare Copy Number Variation in 22q11.2 Deletion Syndrome. Front. Genet. 2022, 13, 812183. [Google Scholar] [CrossRef]
- Xu, B.; Hsu, P.-K.; Stark, K.L.; Karayiorgou, M.; Gogos, J.A. Derepression of a Neuronal Inhibitor Due to miRNA Dysregulation in a Schizophrenia-Related Microdeletion. Cell 2013, 152, 262–275. [Google Scholar] [CrossRef]
- Su, H.; Trombly, M.I.; Chen, J.; Wang, X. Essential and Overlapping Functions for Mammalian Argonautes in microRNA Silencing. Genes Dev. 2009, 23, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Golden, R.J.; Chen, B.; Li, T.; Braun, J.; Manjunath, H.; Chen, X.; Wu, J.; Schmid, V.; Chang, T.-C.; Kopp, F.; et al. An Argonaute Phosphorylation Cycle Promotes microRNA-Mediated Silencing. Nature 2017, 542, 197–202. [Google Scholar] [CrossRef]
- Quévillon Huberdeau, M.; Zeitler, D.M.; Hauptmann, J.; Bruckmann, A.; Fressigné, L.; Danner, J.; Piquet, S.; Strieder, N.; Engelmann, J.C.; Jannot, G.; et al. Phosphorylation of Argonaute Proteins Affects mRNA Binding and Is Essential for microRNA-guided Gene Silencing in Vivo. EMBO J. 2017, 36, 2088–2106. [Google Scholar] [CrossRef]
- Duan, Y.; Li, L.; Panzade, G.P.; Piton, A.; Zinovyeva, A.; Ambros, V. Modeling Neurodevelopmental Disorder-Associated Human AGO1 Mutations in Caenorhabditis elegans Argonaute Alg-1. Proc. Natl. Acad. Sci. USA 2024, 121, e2308255121. [Google Scholar] [CrossRef]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The Biochemical Basis of microRNA Targeting Efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Briskin, D.; Wang, P.Y.; Bartel, D.P. The Biochemical Basis for the Cooperative Action of microRNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 17764–17774. [Google Scholar] [CrossRef]
- Cherone, J.M.; Jorgji, V.; Burge, C.B. Cotargeting among microRNAs in the Brain. Genome Res. 2019, 29, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Kitai, H.; Kato, N.; Ogami, K.; Komatsu, S.; Watanabe, Y.; Yoshino, S.; Koshi, E.; Tsubota, S.; Funahashi, Y.; Maeda, T.; et al. Systematic Characterization of Seed Overlap microRNA Cotargeting Associated with Lupus Pathogenesis. BMC Biol. 2022, 20, 248. [Google Scholar] [CrossRef]
- Hausser, J.; Zavolan, M. Identification and Consequences of miRNA–Target Interactions—Beyond Repression of Gene Expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef]
- Grigelioniene, G.; Suzuki, H.I.; Taylan, F.; Mirzamohammadi, F.; Borochowitz, Z.U.; Ayturk, U.M.; Tzur, S.; Horemuzova, E.; Lindstrand, A.; Weis, M.A.; et al. Gain-of-Function Mutation of microRNA-140 in Human Skeletal Dysplasia. Nat. Med. 2019, 25, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Spengler, R.M.; Grigelioniene, G.; Kobayashi, T.; Sharp, P.A. Deconvolution of Seed and RNA-Binding Protein Crosstalk in RNAi-Based Functional Genomics. Nat. Genet. 2018, 50, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.-T.; Corces, V.G. Enhancer Function: New Insights into the Regulation of Tissue-Specific Gene Expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA Genes to the Core Transcriptional Regulatory Circuitry of Embryonic Stem Cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Mencía, Á.; Modamio-Høybjør, S.; Redshaw, N.; Morín, M.; Mayo-Merino, F.; Olavarrieta, L.; Aguirre, L.A.; del Castillo, I.; Steel, K.P.; Dalmay, T.; et al. Mutations in the Seed Region of Human miR-96 Are Responsible for Nonsyndromic Progressive Hearing Loss. Nat. Genet. 2009, 41, 609–613. [Google Scholar] [CrossRef]
- Lewis, M.A.; Di Domenico, F.; Ingham, N.J.; Prosser, H.M.; Steel, K.P. Hearing Impairment Due to Mir183/96/182 Mutations Suggests Both Loss and Gain of Function Effects. Dis. Model. Mech. 2020, 14, dmm047225. [Google Scholar] [CrossRef]
- Hughes, A.E.; Bradley, D.T.; Campbell, M.; Lechner, J.; Dash, D.P.; Simpson, D.A.; Willoughby, C.E. Mutation Altering the miR-184 Seed Region Causes Familial Keratoconus with Cataract. Am. J. Hum. Genet. 2011, 89, 628–633. [Google Scholar] [CrossRef]
- Iliff, B.W.; Riazuddin, S.A.; Gottsch, J.D. A Single-Base Substitution in the Seed Region of miR-184 Causes EDICT Syndrome. Investig. Ophthalmol. Vis. Sci. 2012, 53, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; Bae, H.A.; Guduric-Fuchs, J.; Rice, A.; Govindarajan, G.; Siddiqui, S.; Abi Farraj, L.; Yip, S.P.; Yap, M.; Das, M.; et al. Mutational Analysis of MIR184 in Sporadic Keratoconus and Myopia. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5266–5272. [Google Scholar] [CrossRef] [PubMed]
- Conte, I.; Hadfield, K.D.; Barbato, S.; Carrella, S.; Pizzo, M.; Bhat, R.S.; Carissimo, A.; Karali, M.; Porter, L.F.; Urquhart, J.; et al. MiR-204 Is Responsible for Inherited Retinal Dystrophy Associated with Ocular Coloboma. Proc. Natl. Acad. Sci. USA 2015, 112, E3236–E3245. [Google Scholar] [CrossRef]
- de Pontual, L.; Yao, E.; Callier, P.; Faivre, L.; Drouin, V.; Cariou, S.; Van Haeringen, A.; Geneviève, D.; Goldenberg, A.; Oufadem, M.; et al. Germline Deletion of the miR-17-92 Cluster Causes Growth and Skeletal Defects in Humans. Nat. Genet. 2011, 43, 1026–1030. [Google Scholar] [CrossRef]
- Henrion-Caude, A.; Girard, M.; Amiel, J. MicroRNAs in Genetic Disease: Rethinking the Dosage. Curr. Gene Ther. 2012, 12, 292–300. [Google Scholar] [CrossRef]
- Miyakawa, K.; Miyashita, N.; Horie, M.; Terasaki, Y.; Tanaka, H.; Urushiyama, H.; Fukuda, K.; Okabe, Y.; Ishii, T.; Kuwahara, N.; et al. ASCL1 Regulates Super-enhancer-associated miRNAs to Define Molecular Subtypes of Small Cell Lung Cancer. Cancer Sci. 2022, 113, 3932–3946. [Google Scholar] [CrossRef]
- Luciano, D.J.; Mirsky, H.; Vendetti, N.J.; Maas, S. RNA Editing of a miRNA Precursor. RNA 2004, 10, 1174–1177. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA Processing and Expression through RNA Editing by ADAR Deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.; Player, C.; Axtell, M.J.; Lee, W.; Nusbaum, C.; Ge, H.; Bartel, D.P. Large-Scale Sequencing Reveals 21U-RNAs and Additional microRNAs and Endogenous siRNAs in C. elegans. Cell 2006, 127, 1193–1207. [Google Scholar] [CrossRef]
- Burroughs, A.M.; Ando, Y.; De Hoon, M.J.L.; Tomaru, Y.; Nishibu, T.; Ukekawa, R.; Funakoshi, T.; Kurokawa, T.; Suzuki, H.; Hayashizaki, Y.; et al. A Comprehensive Survey of 3′ Animal miRNA Modification Events and a Possible Role for 3′ Adenylation in Modulating miRNA Targeting Effectiveness. Genome Res. 2010, 20, 1398–1410. [Google Scholar] [CrossRef]
- Seok, H.; Lee, H.; Lee, S.; Ahn, S.H.; Lee, H.-S.; Kim, G.-W.D.; Peak, J.; Park, J.; Cho, Y.K.; Jeong, Y.; et al. Position-Specific Oxidation of miR-1 Encodes Cardiac Hypertrophy. Nature 2020, 584, 279–285. [Google Scholar] [CrossRef] [PubMed]
- del Valle-Morales, D.; Le, P.; Saviana, M.; Romano, G.; Nigita, G.; Nana-Sinkam, P.; Acunzo, M. The Epitranscriptome in miRNAs: Crosstalk, Detection, and Function in Cancer. Genes 2022, 13, 1289. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Knouf, E.C.; Parkin, R.K.; Fritz, B.R.; Lin, D.W.; Dennis, L.M.; Krouse, M.A.; Webster, P.J.; Tewari, M. Post-Transcriptional Generation of miRNA Variants by Multiple Nucleotidyl Transferases Contributes to miRNA Transcriptome Complexity. Genome Res. 2011, 21, 1450–1461. [Google Scholar] [CrossRef]
- Newman, M.A.; Mani, V.; Hammond, S.M. Deep Sequencing of microRNA Precursors Reveals Extensive 3′ End Modification. RNA 2011, 17, 1795–1803. [Google Scholar] [CrossRef]
- Chiang, H.R.; Schoenfeld, L.W.; Ruby, J.G.; Auyeung, V.C.; Spies, N.; Baek, D.; Johnston, W.K.; Russ, C.; Luo, S.; Babiarz, J.E.; et al. Mammalian microRNAs: Experimental Evaluation of Novel and Previously Annotated Genes. Genes. Dev. 2010, 24, 992–1009. [Google Scholar] [CrossRef]
- Ameres, S.L.; Horwich, M.D.; Hung, J.-H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA–Directed Trimming and Tailing of Small Silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective Blockade of microRNA Processing by Lin-28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef]
- Heo, I.; Joo, C.; Cho, J.; Ha, M.; Han, J.; Kim, V.N. Lin28 Mediates the Terminal Uridylation of Let-7 Precursor MicroRNA. Mol. Cell 2008, 32, 276–284. [Google Scholar] [CrossRef]
- Piskounova, E.; Viswanathan, S.R.; Janas, M.; LaPierre, R.J.; Daley, G.Q.; Sliz, P.; Gregory, R.I. Determinants of MicroRNA Processing Inhibition by the Developmentally Regulated RNA-Binding Protein Lin28. J. Biol. Chem. 2008, 283, 21310–21314. [Google Scholar] [CrossRef]
- Heo, I.; Joo, C.; Kim, Y.-K.; Ha, M.; Yoon, M.-J.; Cho, J.; Yeom, K.-H.; Han, J.; Kim, V.N. TUT4 in Concert with Lin28 Suppresses MicroRNA Biogenesis through Pre-MicroRNA Uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef]
- Hagan, J.P.; Piskounova, E.; Gregory, R.I. Lin28 Recruits the TUTase Zcchc11 to Inhibit Let-7 Maturation in Mouse Embryonic Stem Cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.E.; Chang, H.-M.; Piskounova, E.; Gregory, R.I. Lin28-Mediated Control of Let-7 microRNA Expression by Alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 2012, 18, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Hojo, H.; Suzuki, T. Destabilization of microRNAs in Human Cells by 3′ Deadenylation Mediated by PARN and CUGBP1. Nucleic Acids Res. 2015, 43, 7521–7534. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Yashiro, Y.; Noda, Y.; Ogami, K.; Yamagishi, R.; Okada, S.; Hoshino, S.; Suzuki, T. The RNA-Binding Protein QKI-7 Recruits the Poly(A) Polymerase GLD-2 for 3′ Adenylation and Selective Stabilization of microRNA-122. J. Biol. Chem. 2020, 295, 390–402. [Google Scholar] [CrossRef]
- Chang, H.-M.; Triboulet, R.; Thornton, J.E.; Gregory, R.I. A Role for the Perlman Syndrome Exonuclease Dis3l2 in the Lin28–Let-7 Pathway. Nature 2013, 497, 244–248. [Google Scholar] [CrossRef]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.-J.; Park, J.-E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-Uridylation of Pre-MicroRNA as a Key Step in the Biogenesis of Group II Let-7 MicroRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef]
- Jones, M.R.; Quinton, L.J.; Blahna, M.T.; Neilson, J.R.; Fu, S.; Ivanov, A.R.; Wolf, D.A.; Mizgerd, J.P. Zcchc11-Dependent Uridylation of microRNA Directs Cytokine Expression. Nat. Cell Biol. 2009, 11, 1157–1163. [Google Scholar] [CrossRef]
- Katoh, T.; Sakaguchi, Y.; Miyauchi, K.; Suzuki, T.; Kashiwabara, S.; Baba, T.; Suzuki, T. Selective Stabilization of Mammalian microRNAs by 3′ Adenylation Mediated by the Cytoplasmic Poly(A) Polymerase GLD-2. Genes. Dev. 2009, 23, 433–438. [Google Scholar] [CrossRef]
- Boele, J.; Persson, H.; Shin, J.W.; Ishizu, Y.; Newie, I.S.; Søkilde, R.; Hawkins, S.M.; Coarfa, C.; Ikeda, K.; Takayama, K.; et al. PAPD5-Mediated 3′ Adenylation and Subsequent Degradation of miR-21 Is Disrupted in Proliferative Disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11467–11472. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.; Yu, S.; Lee, Y.-Y.; Park, J.; Choi, R.J.; Yoon, S.-J.; Kang, S.-G.; Kim, V.N. A Mechanism for microRNA Arm Switching Regulated by Uridylation. Mol. Cell 2020, 78, 1224–1236.e5. [Google Scholar] [CrossRef]
- Yang, A.; Bofill-De Ros, X.; Shao, T.-J.; Jiang, M.; Li, K.; Villanueva, P.; Dai, L.; Gu, S. 3′ Uridylation Confers miRNAs with Non-Canonical Target Repertoires. Mol. Cell 2019, 75, 511–522.e4. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-Regulation of a Host MicroRNA by a Herpesvirus Saimiri Noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.Y.; Kingston, E.R.; Kleaveland, B.; Lin, D.H.; Stubna, M.W.; Bartel, D.P. The ZSWIM8 Ubiquitin Ligase Mediates Target-Directed microRNA Degradation. Science 2020, 370, eabc9359. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; LaVigne, C.A.; Jones, B.T.; Zhang, H.; Gillett, F.; Mendell, J.T. A Ubiquitin Ligase Mediates Target-Directed microRNA Decay Independently of Tailing and Trimming. Science 2020, 370, eabc9546. [Google Scholar] [CrossRef]
- Jones, B.T.; Han, J.; Zhang, H.; Hammer, R.E.; Evers, B.M.; Rakheja, D.; Acharya, A.; Mendell, J.T. Target-Directed microRNA Degradation Regulates Developmental microRNA Expression and Embryonic Growth in Mammals. Genes. Dev. 2023, 37, 661–674. [Google Scholar] [CrossRef]
- Haas, G.; Cetin, S.; Messmer, M.; Chane-Woon-Ming, B.; Terenzi, O.; Chicher, J.; Kuhn, L.; Hammann, P.; Pfeffer, S. Identification of Factors Involved in Target RNA-Directed microRNA Degradation. Nucleic Acids Res. 2016, 44, 2873–2887. [Google Scholar] [CrossRef]
- Yang, A.; Shao, T.-J.; Bofill-De Ros, X.; Lian, C.; Villanueva, P.; Dai, L.; Gu, S. AGO-Bound Mature miRNAs Are Oligouridylated by TUTs and Subsequently Degraded by DIS3L2. Nat. Commun. 2020, 11, 2765. [Google Scholar] [CrossRef]
- Wu, P.-H.; Zamore, P.D. To Degrade a MicroRNA, Destroy Its Argonaute Protein. Mol. Cell 2021, 81, 223–225. [Google Scholar] [CrossRef]
- de la Mata, M.; Gaidatzis, D.; Vitanescu, M.; Stadler, M.B.; Wentzel, C.; Scheiffele, P.; Filipowicz, W.; Großhans, H. Potent Degradation of Neuronal miRNAs Induced by Highly Complementary Targets. EMBO Rep. 2015, 16, 500–511. [Google Scholar] [CrossRef]
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 2018, 174, 350–362.e17. [Google Scholar] [CrossRef]
- Snoek, B.C.; Babion, I.; Koppers-Lalic, D.; Pegtel, D.M.; Steenbergen, R.D. Altered microRNA Processing Proteins in HPV-Induced Cancers. Curr. Opin. Virol. 2019, 39, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hilcenko, C.; Simpson, P.J.; Finch, A.J.; Bowler, F.R.; Churcher, M.J.; Jin, L.; Packman, L.C.; Shlien, A.; Campbell, P.; Kirwan, M.; et al. Aberrant 3′ Oligoadenylation of Spliceosomal U6 Small Nuclear RNA in Poikiloderma with Neutropenia. Blood 2013, 121, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E. Evolution of microRNA Diversity and Regulation in Animals. Nat. Rev. Genet. 2011, 12, 846–860. [Google Scholar] [CrossRef]
- Quah, S.; Subramanian, G.; Tan, J.S.L.; Utami, K.H.; Sampath, P. MicroRNAs: A Symphony Orchestrating Evolution and Disease Dynamics. Trends Mol. Med. 2025, 31, 21–35. [Google Scholar] [CrossRef]
- Hertel, J.; Lindemeyer, M.; Missal, K.; Fried, C.; Tanzer, A.; Flamm, C.; Hofacker, I.L.; Stadler, P.F. Students of Bioinformatics Computer Labs 2004 and 2005 The Expansion of the Metazoan microRNA Repertoire. BMC Genom. 2006, 7, 25. [Google Scholar] [CrossRef]
- Berezikov, E.; Thuemmler, F.; van Laake, L.W.; Kondova, I.; Bontrop, R.; Cuppen, E.; Plasterk, R.H.A. Diversity of microRNAs in Human and Chimpanzee Brain. Nat. Genet. 2006, 38, 1375–1377. [Google Scholar] [CrossRef]
- Todorov, H.; Weißbach, S.; Schlichtholz, L.; Mueller, H.; Hartwich, D.; Gerber, S.; Winter, J. Stage-Specific Expression Patterns and Co-Targeting Relationships among miRNAs in the Developing Mouse Cerebral Cortex. Commun. Biol. 2024, 7, 1366. [Google Scholar] [CrossRef]
- Miura, P.; Shenker, S.; Andreu-Agullo, C.; Westholm, J.O.; Lai, E.C. Widespread and Extensive Lengthening of 3′ UTRs in the Mammalian Brain. Genome Res. 2013, 23, 812–825. [Google Scholar] [CrossRef]
- Thomas, K.T.; Vermare, A.; Egleston, S.O.; Wang, Y.-D.; Mishra, A.; Lin, T.; Peng, J.; Zakharenko, S.S. MicroRNA 3′ Ends Shorten during Adolescent Brain Maturation. Front. Mol. Neurosci. 2023, 16, 1168695. [Google Scholar] [CrossRef]
- Krol, J.; Busskamp, V.; Markiewicz, I.; Stadler, M.B.; Ribi, S.; Richter, J.; Duebel, J.; Bicker, S.; Fehling, H.J.; Schübeler, D.; et al. Characterizing Light-Regulated Retinal MicroRNAs Reveals Rapid Turnover as a Common Property of Neuronal MicroRNAs. Cell 2010, 141, 618–631. [Google Scholar] [CrossRef]
- Vattathil, S.M.; Gerasimov, E.S.; Canon, S.M.; Lori, A.; Tan, S.S.M.; Kim, P.J.; Liu, Y.; Lai, E.C.; Bennett, D.A.; Wingo, T.S.; et al. Mapping the microRNA Landscape in the Older Adult Brain and Its Genetic Contribution to Neuropsychiatric Conditions. Nat. Aging 2024, 5, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.P.; Bruse, S.E.; David-Rus, R.; Buyske, S.; Brzustowicz, L.M. Altered MicroRNA Expression Profiles in Postmortem Brain Samples from Individuals with Schizophrenia and Bipolar Disorder. Biol. Psychiatry 2011, 69, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Y.; Liu, X.; Wu, S.; Wang, M.; Turowski, S.G.; Spernyak, J.A.; Tracz, A.; Abdelaal, A.M.; Sudarshan, K.; et al. Developing Folate-Conjugated miR-34a Therapeutic for Prostate Cancer: Challenges and Promises. Int. J. Mol. Sci. 2024, 25, 2123. [Google Scholar] [CrossRef]
- Abdelaal, A.M.; Sohal, I.S.; Iyer, S.G.; Sudarshan, K.; Orellana, E.A.; Ozcan, K.E.; dos Santos, A.P.; Low, P.S.; Kasinski, A.L. Selective Targeting of Chemically Modified miR-34a to Prostate Cancer Using a Small Molecule Ligand and an Endosomal Escape Agent. Mol. Ther. Nucleic Acids 2024, 35, 102193. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: MicroRNAs Can Up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Vasudevan, S.; Steitz, J.A. AU-Rich-Element-Mediated Upregulation of Translation by FXR1 and Argonaute 2. Cell 2007, 128, 1105–1118. [Google Scholar] [CrossRef]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef]
- Schult, P.; Roth, H.; Adams, R.L.; Mas, C.; Imbert, L.; Orlik, C.; Ruggieri, A.; Pyle, A.M.; Lohmann, V. microRNA-122 Amplifies Hepatitis C Virus Translation by Shaping the Structure of the Internal Ribosomal Entry Site. Nat. Commun. 2018, 9, 2613. [Google Scholar] [CrossRef]
- Mengardi, C.; Limousin, T.; Ricci, E.P.; Soto-Rifo, R.; Decimo, D.; Ohlmann, T. microRNAs Stimulate Translation Initiation Mediated by HCV-like IRESes. Nucleic Acids Res. 2017, 45, 4810–4824. [Google Scholar] [CrossRef]
- Wakiyama, M.; Ogami, K.; Iwaoka, R.; Aoki, K.; Hoshino, S. MicroRNP-Mediated Translational Activation of Nonadenylated mRNAs in a Mammalian Cell-Free System. Genes Cells 2018, 23, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, X.; Wang, F.; Zhou, L.; Yin, Z.; Fan, J.; Nie, X.; Wang, P.; Fu, X.-D.; Chen, C.; et al. MicroRNA-21 Lowers Blood Pressure in Spontaneous Hypertensive Rats by Upregulating Mitochondrial Translation. Circulation 2016, 134, 734–751. [Google Scholar] [CrossRef] [PubMed]
- Jame-Chenarboo, F.; Reyes, J.N.; Twells, N.M.; Ng, H.H.; Macdonald, D.; Hernando, E.; Mahal, L.K. Screening the Human miRNA Interactome Reveals Coordinated Up-Regulation in Melanoma, Adding Bidirectional Regulation to miRNA Networks. Sci. Adv. 2025, 11, eadr0277. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.I.A.; Truesdell, S.S.; Lee, S.; Kollu, S.; Classon, A.; Boukhali, M.; Jain, E.; Mortensen, R.D.; Yanagiya, A.; Sadreyev, R.I.; et al. A Specialized Mechanism of Translation Mediated by FXR1a-Associated MicroRNP in Cellular Quiescence. Mol. Cell 2016, 61, 760–773. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Disease | Phenotypes | References | |
|---|---|---|---|---|
| miRNA biogenesis pathway | DROSHA | Wilms tumor (point mutation) | Genitourinary tract cancer | [40,41,42,43] |
| DGCR8 | DiGeorge syndrome (22q11.2 deletion) | Congenital heart defects, developmental delay, behavioral and cognitive deficits, learning disabilities, immunodeficiency, hypocalcemia, characteristic facial features, and schizophrenia | [44,45,46,47,48,49] | |
| Wilms tumor (point mutation) | Genitourinary tract cancer | [42,43] | ||
| Tumor susceptibility syndrome (point mutation, loss-of-heterozygosity) | Euthyroid multinodular goiter (MNG) with schwannomatosis | [50] | ||
| DICER1 | DICER1 Syndrome (point mutation, loss-of-heterozygosity) | Pleuropulmonary blastoma, ovarian Sertoli–Leydig cell tumor, cystic nephroma, embryonal rhabdomyosarcoma, and multinodular goiter | [51,52,53,54,55] | |
| Wilms tumor (point mutation) | Genitourinary tract cancer | [40,41,56] | ||
| AGO1 AGO2 | AGO syndrome (1p34.3 deletion, point mutation) | Psychomotor developmental delays, neurological development disorder (NDD), intellectual disability (ID), delayed motor development, impaired speech and receptive language skills, and autism spectrum disorder (ASD) | [57,58,59,60,61,62,63] | |
| miRNA 3′-end processing | PARN | Dyskeratosis congenita (DC) (point mutation, deletion) | Telomere shortening, nail atrophy, oral leukoplakia, cutaneous hyperpigmentation, pulmonary fibrosis, bone marrow failure, and cancer | [64,65,66,67,68] |
| USB1 | Poikiloderma with neutropenia (PN) (point mutation, deletion) | Poikiloderma, decrease in neutrophils, bone marrow failure, and cancer | [68,69,70,71] |
| miRNA Gene | Disease | Phenotypes | References |
|---|---|---|---|
| miR-96 | Non-syndromic hearing impairment (point mutation) | Progressive hearing loss | [98,99] |
| miR-184 | EDICT syndrome (point mutation) | Endothelial dystrophy, iris hypoplasia, congenital cataract, and stromal thinning | [100,101,102] |
| miR-204 | Ocular coloboma (point mutation) | Retinal dystrophy | [103] |
| miR-140 | Skeletal dysplasia (spondyloepiphyseal dysplasia (SED) MIR140 type Nishimura) (point mutation, SE-associated) | Disproportionately short stature, and spondylar and epiphyseal abnormalities | [91] |
| MIR17HG | Feingold syndrome (13q31.3 deletion) | Microcephaly, short stature, and digital abnormalities | [104,105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seida, M.; Ogami, K.; Yoshino, S.; Suzuki, H.I. Fine Regulation of MicroRNAs in Gene Regulatory Networks and Pathophysiology. Int. J. Mol. Sci. 2025, 26, 2861. https://doi.org/10.3390/ijms26072861
Seida M, Ogami K, Yoshino S, Suzuki HI. Fine Regulation of MicroRNAs in Gene Regulatory Networks and Pathophysiology. International Journal of Molecular Sciences. 2025; 26(7):2861. https://doi.org/10.3390/ijms26072861
Chicago/Turabian StyleSeida, Mayu, Koichi Ogami, Seiko Yoshino, and Hiroshi I. Suzuki. 2025. "Fine Regulation of MicroRNAs in Gene Regulatory Networks and Pathophysiology" International Journal of Molecular Sciences 26, no. 7: 2861. https://doi.org/10.3390/ijms26072861
APA StyleSeida, M., Ogami, K., Yoshino, S., & Suzuki, H. I. (2025). Fine Regulation of MicroRNAs in Gene Regulatory Networks and Pathophysiology. International Journal of Molecular Sciences, 26(7), 2861. https://doi.org/10.3390/ijms26072861