
Structure and Optical Properties of New 2-N-Phenylamino-methyl-nitro-pyridine Isomers



 , , and
, , and
Abstract

1. Introduction
2. Results and Discussion
2.1. Structural Characterization
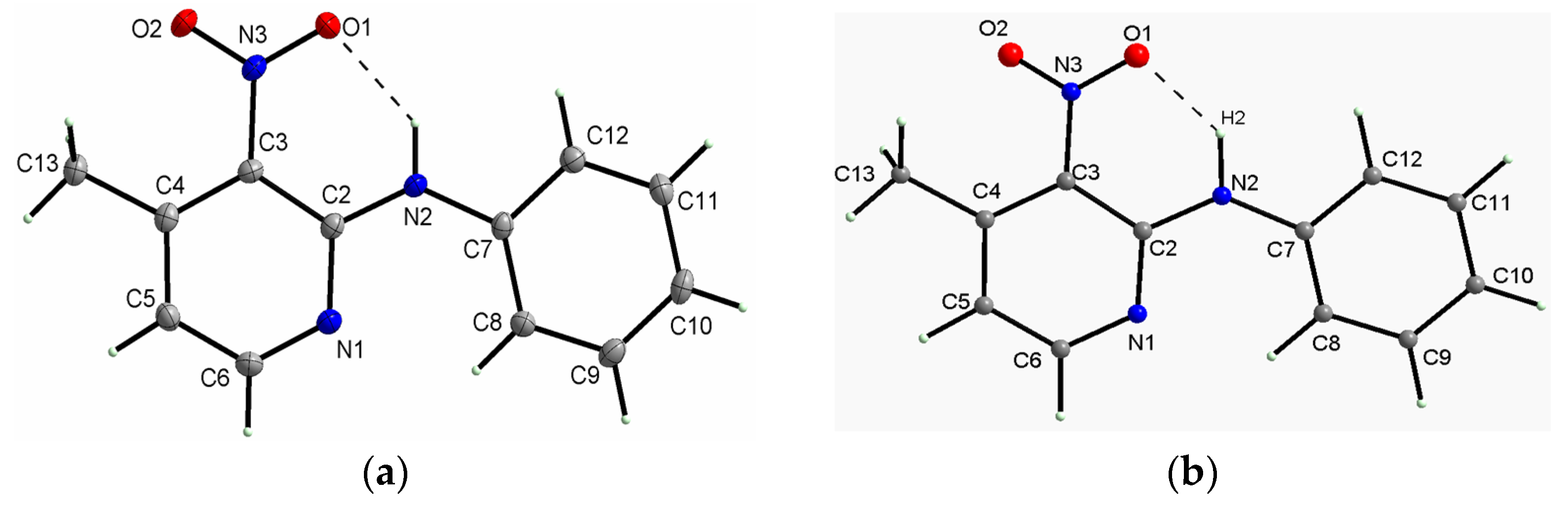
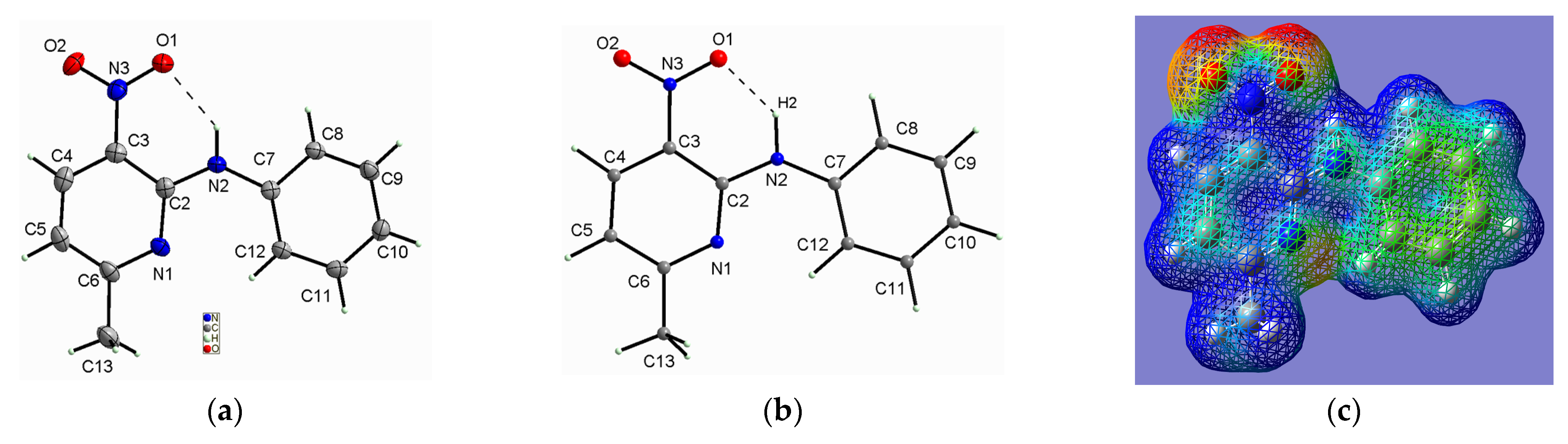
2.1.1. Description of the Structure of 2-N-Phenylamino-3-nitro-4-methylpyridine
2.1.2. Description of the Structure of 2-N-Phenylamino-3-nitro-6-methylpyridine
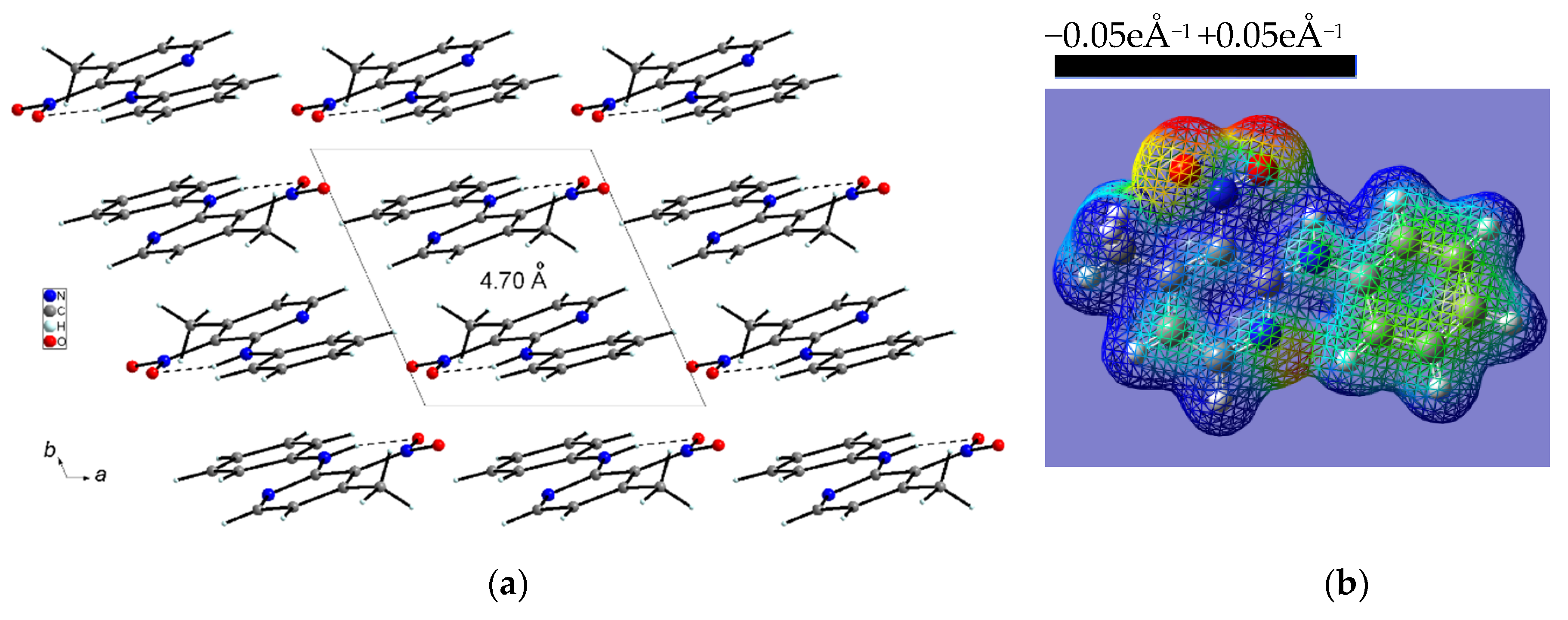
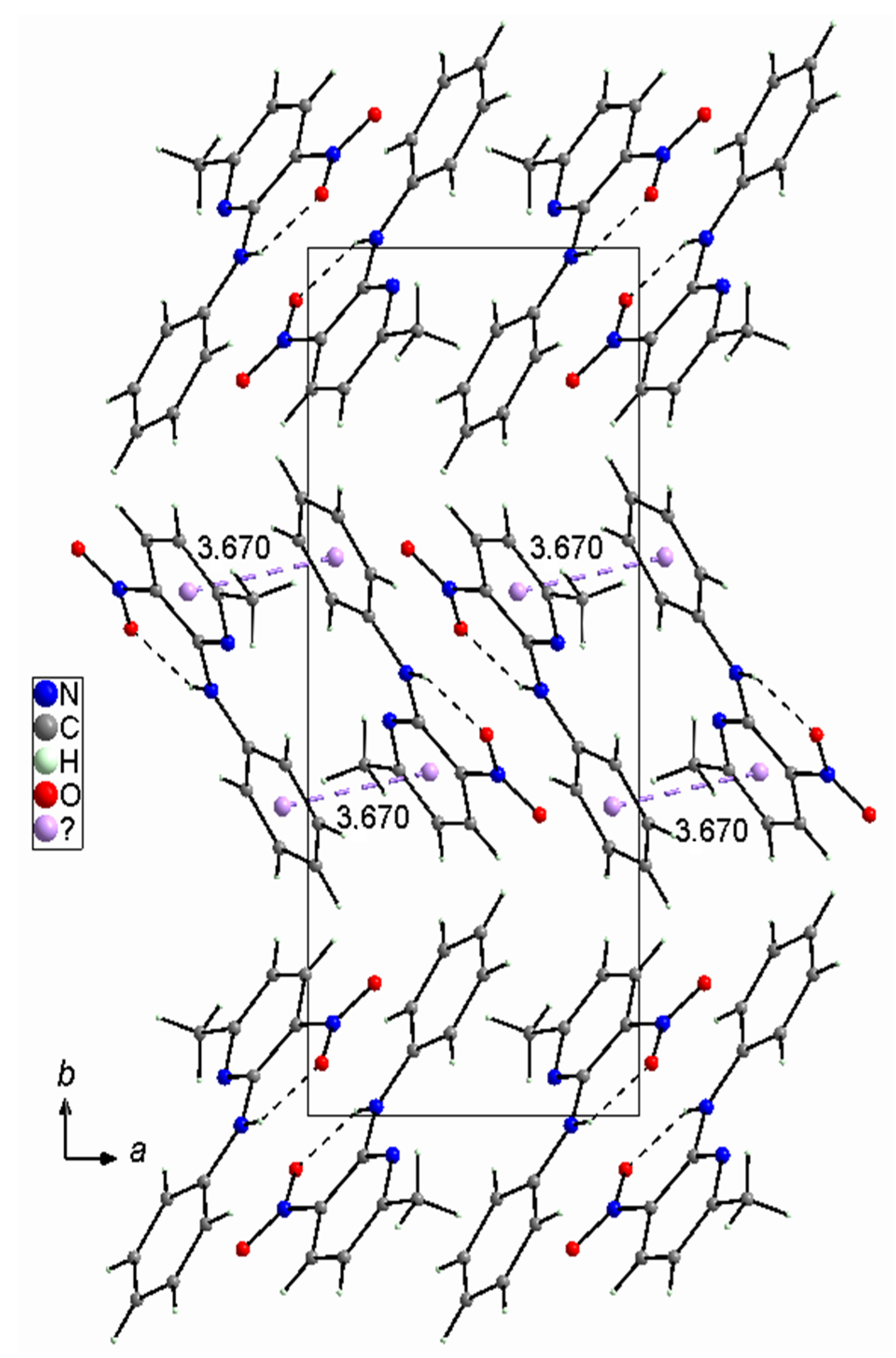
2.1.3. Comparison of the Structures of PA3N4MP and PA3N6MP
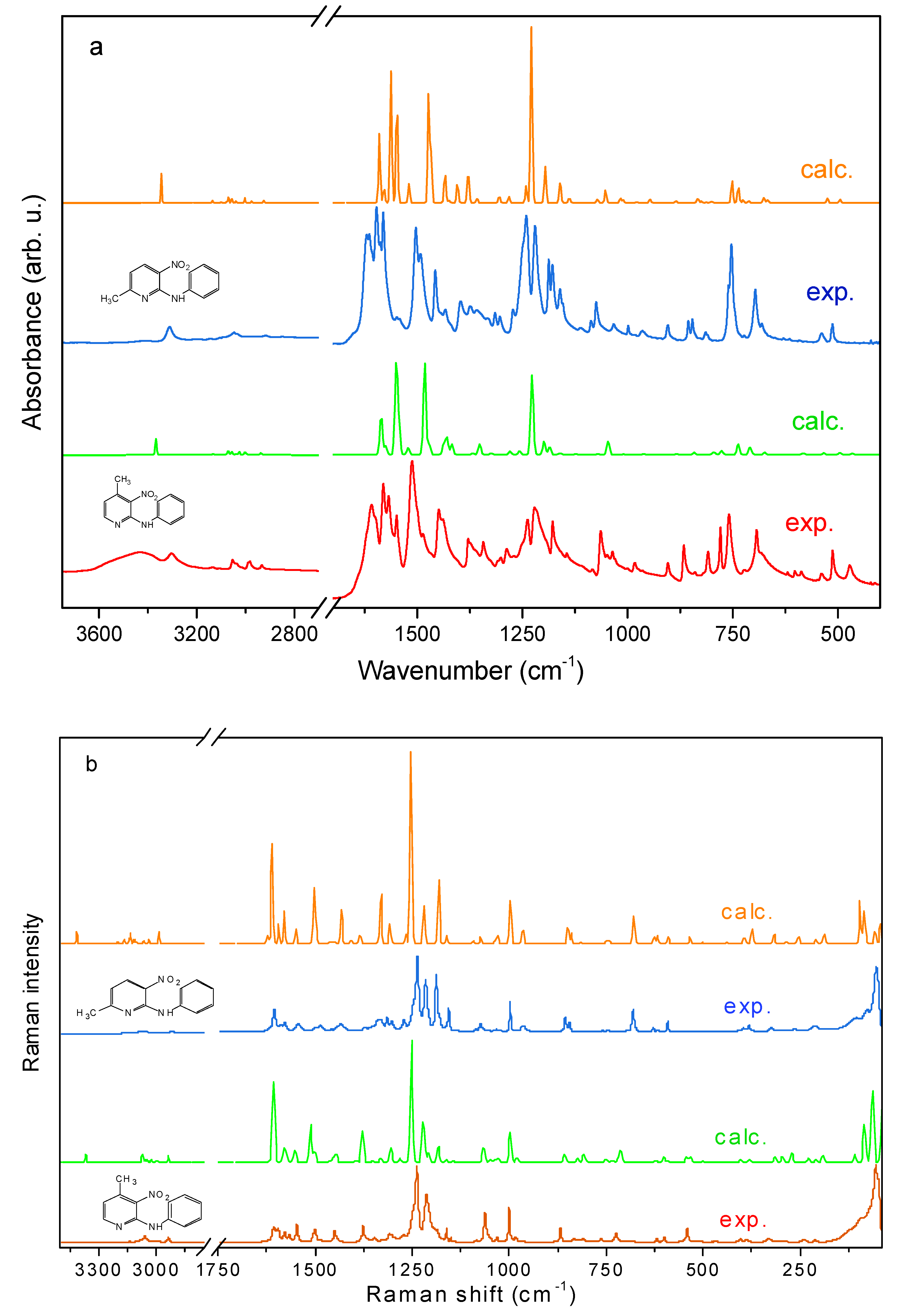
2.2. Vibration Characterization
2.2.1. Vibrational Spectra
2.2.2. Vibrational Modes of Pyridine and Phenyl Systems
- Localized modes arising from discrete atomic displacements, including the following:
- ○
- N–H stretching (ν(NH));
- ○
- Aromatic C–H stretching (ν(CH));
- ○
- Out-of-plane C–H bending (γ(CH));
- ○
- Methyl group vibrations (ν(CH3;), δ(CH3;), ρ(CH3;)).
- Collective vibrational modes involving coupled displacements across multiple atomic coordinates. The following ranges are proposed as the pyridine ring vibrations for derivatives PA3N4MP and PA3N6MP (in parenthesis), respectively: ν(CH): 3036–3056, 3130 (3047–3146); ν(ϕ): 1148–1486 (1458–1491); ν(ϕ) + δ(CNH)ϕ: 1579–1596 (1580–1596); ν(ϕ) + δ(CϕNCΘ): 1500–1548 (1500–1548); δ(ϕ) and δ(ϕ) + δ(CH): 1238–1286 (1236–1271); δ(CH)ϕ: 1048–1063. 810 (1073, 813); τ(ϕ): 600 (592); γ(ϕ): 536–541 (534–536) and δ(ϕ) + ν(Cϕ-NO2): 386 (394–395) cm−1.
2.2.3. Methyl Group Vibrational Modes
- Asymmetric stretching (νas(CH3;)): 2981–3005 cm−1 (2980–3006 cm−1);
- Symmetric stretching (νs(CH3;)): 2932–2935 cm−1 (2914–2915 cm−1);
- Asymmetric bending (δas(CH3;)): 1429–1437 cm−1 (1430–1434 cm−1);
- Symmetric bending (δs(CH3;)): 1326–1278 cm−1 (1315–1373 cm−1);
- In-plane rocking (ρ(CH3;)): 1001–1027 cm−1 (1002–1025 cm−1);
- Torsional modes (τ(CH3;)): ~270 cm−1 (264–278 cm−1).
2.2.4. Nitro Group Vibrations
- Stretching vibrations:
- ○
- Asymmetric (νas(NO2));
- ○
- Symmetric (νs(NO2));
- ○
- C–N linkage stretching (ν(C–NO2)).
- In-plane deformations:
- ○
- Bending (δ(NO2));
- ○
- Rocking (ρ(NO2));
- ○
- Pyridine ring–NO2 bending (δ(φ–NO2)).
- Out-of-plane motions:
- ○
- Wagging (ω(NO2));
- ○
- Twisting (τ(NO2));
- ○
- Pyridine ring–NO2 torsional (γ(φ–NO2))
- νas(NO2): 1378–1567 cm−1 (1434–1578 cm−1);
- νs(NO2): 1162–1378 cm−1 (1156–1373 cm−1);
- Scissoring (δ(NO2)): 833–845 cm−1;
- Wagging (ω(NO2)): 720–725 cm−1.
2.2.5. Vibrations of the Amino Bridge
- PA3N4MP: 3433 cm−1 and 3304 cm−1;
- PA3N6MP: 3430 cm−1 and 3311 cm−1.
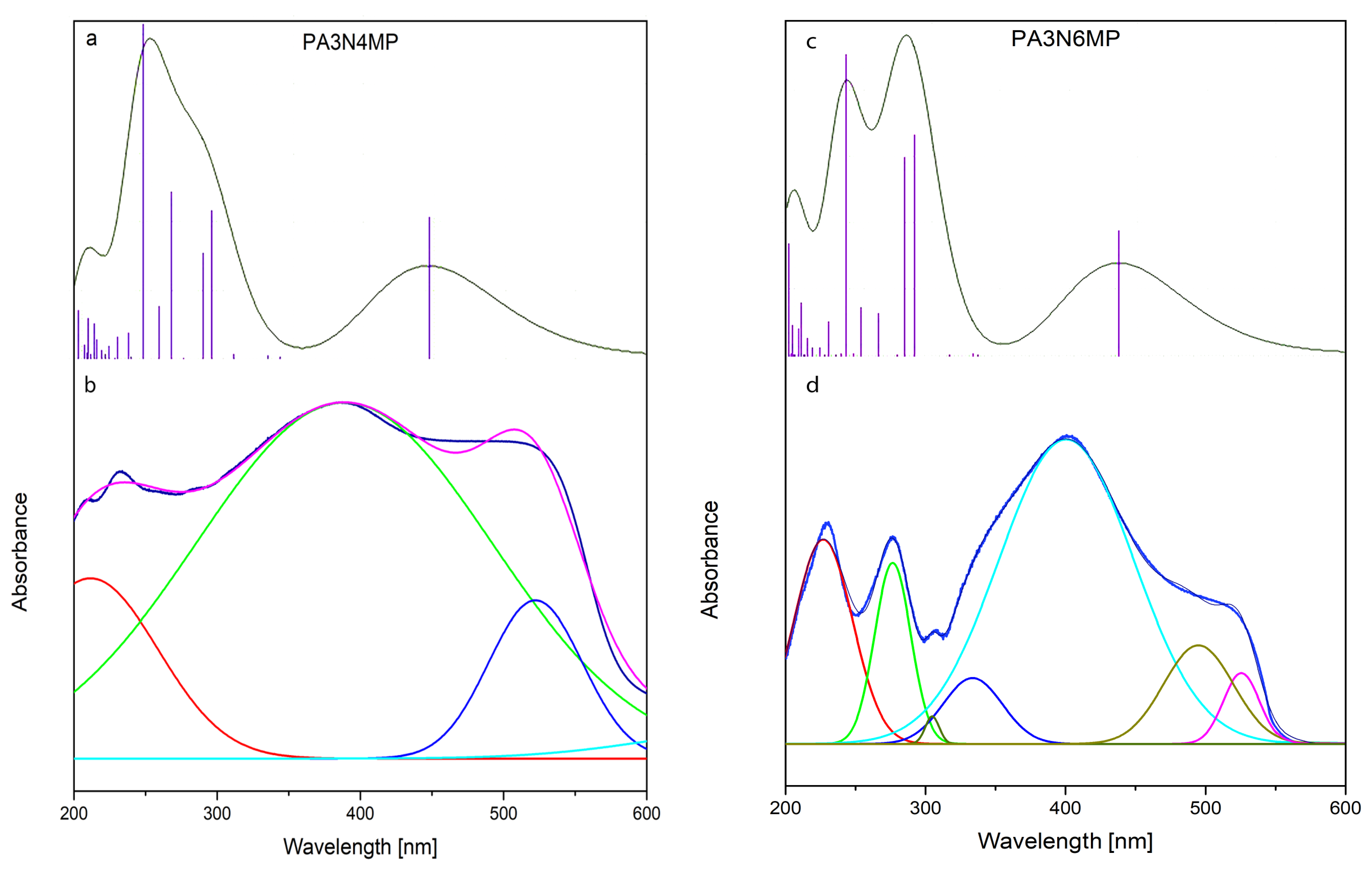
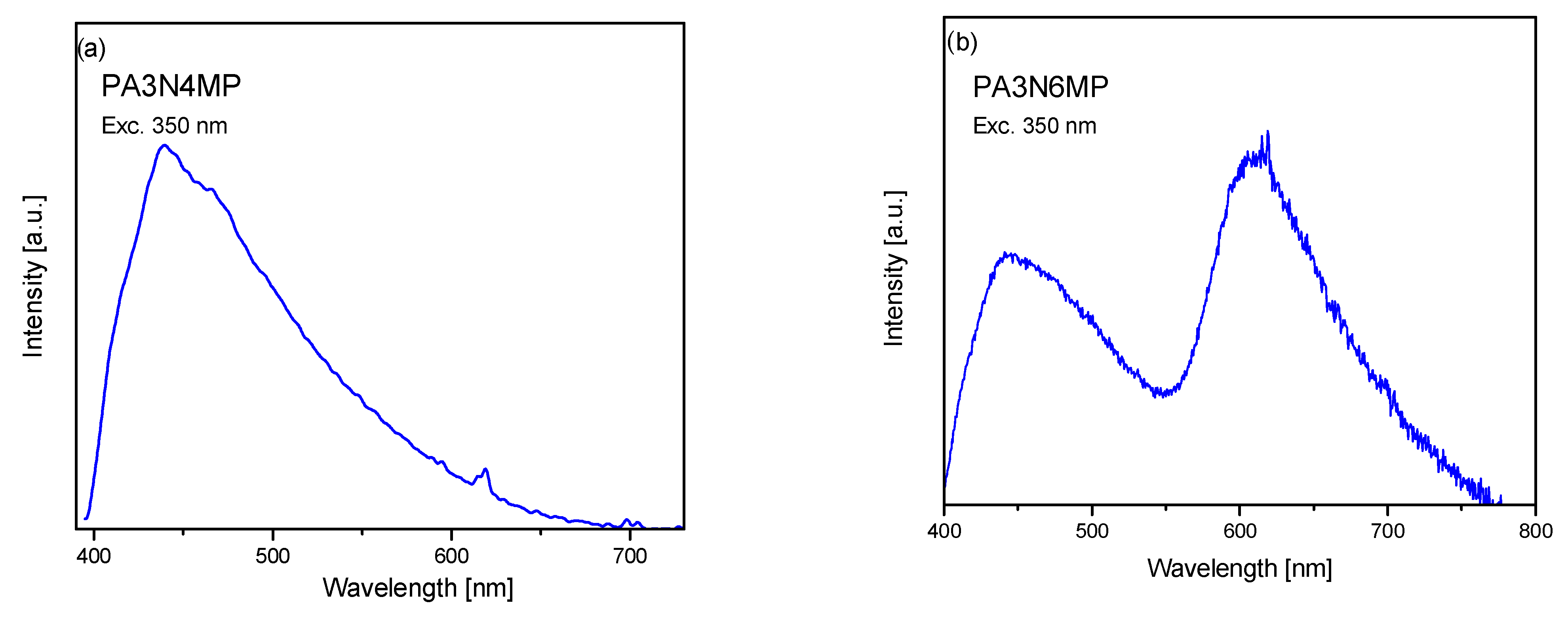
2.3. Electron Reflectance and Emission Spectra
2.4. 13C and 1H NMR Spectra Measurements
- Pyridine ring: five unique carbons;
- methyl group: one resonance;
- phenyl ring: four signals (with two symmetry-equivalent carbon pairs).
3. Materials and Methods
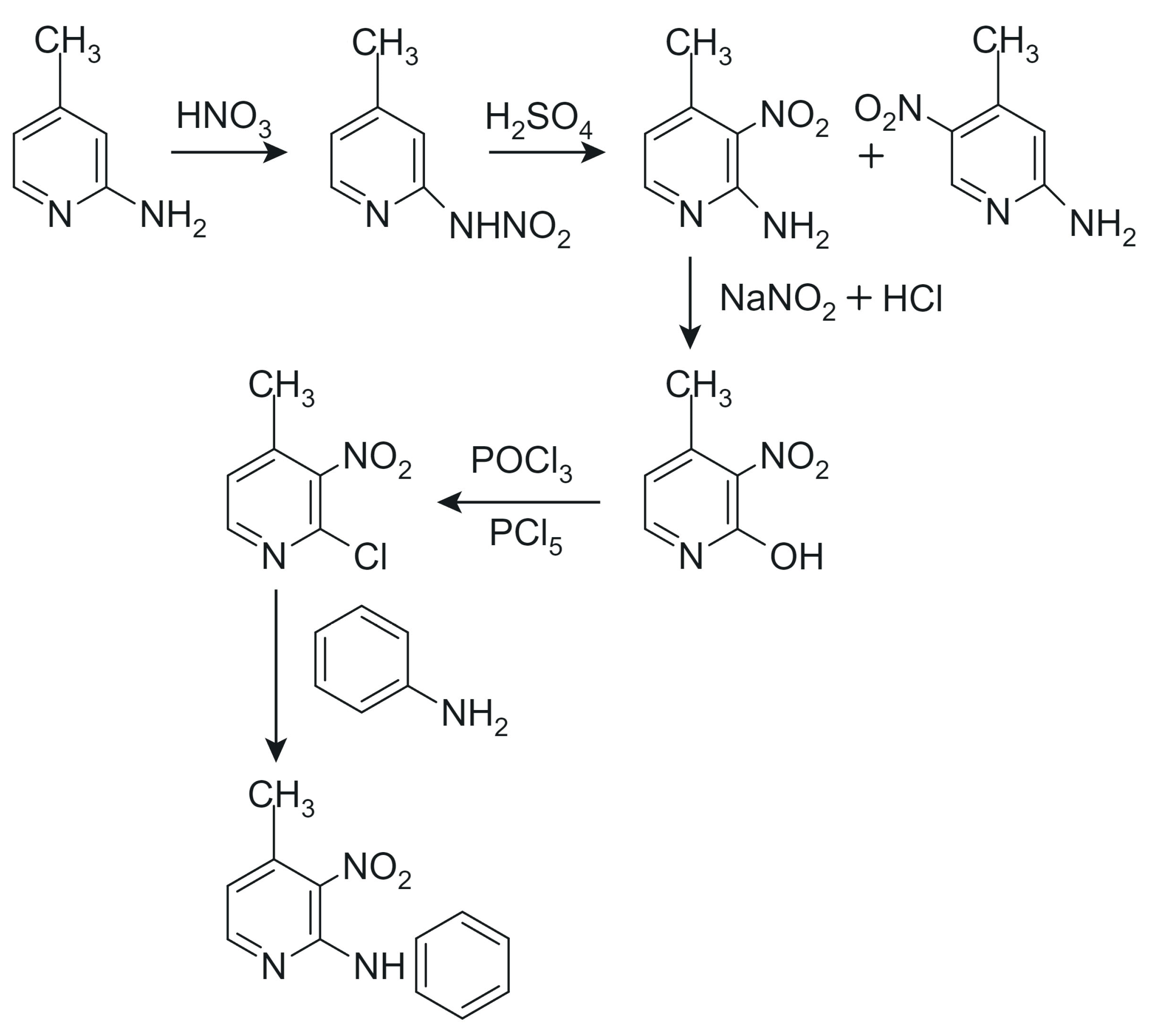
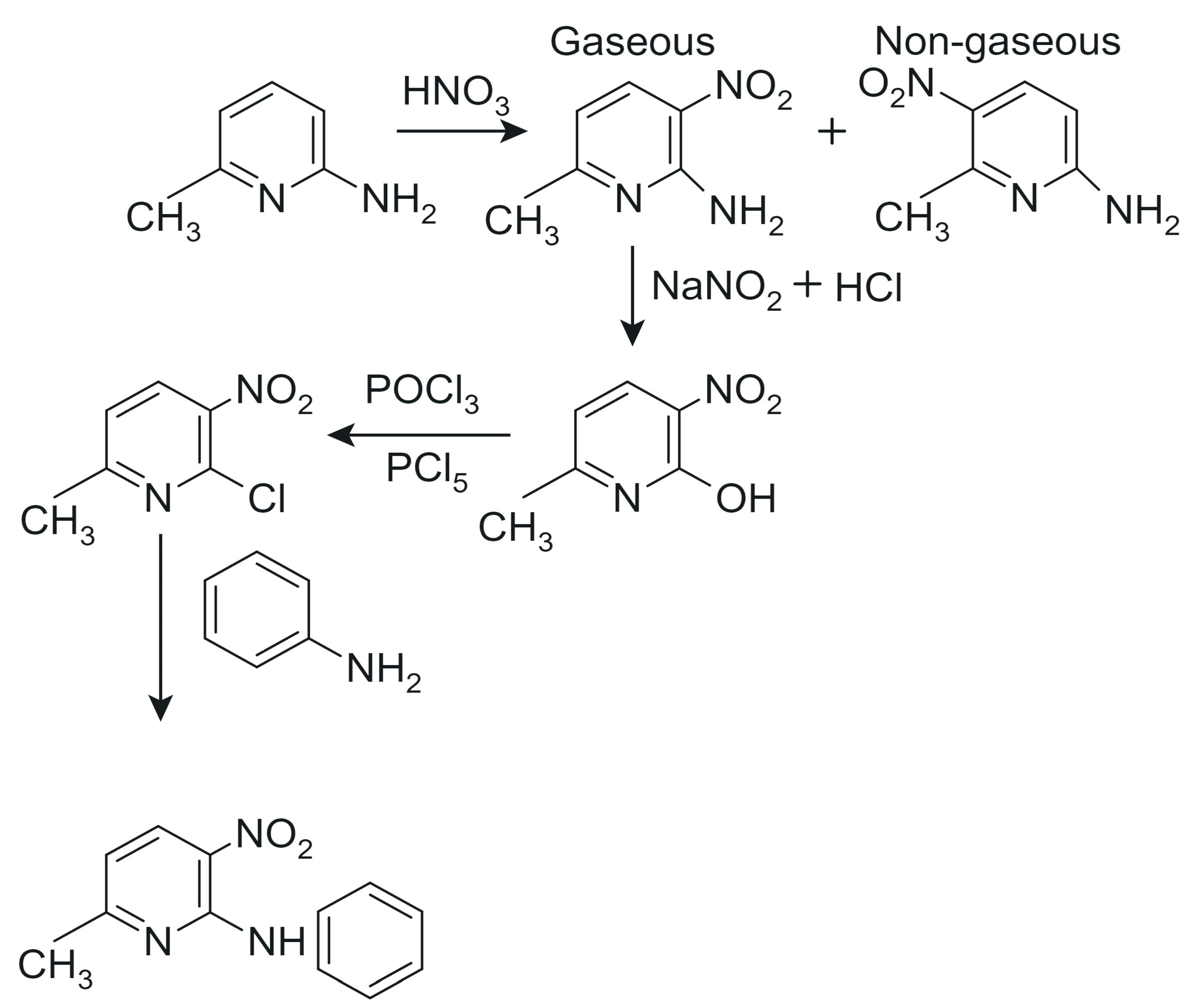
3.1. Synthesis
- 2-N-phenylamino-3-nitro-4-methylpyridine
- 2-N-phenylamino-3-nitro-6-methylpyridine
- 2-N-phenylamino-3-nitro-4-methylpyridine
- Chemical formula: C12H11N3O2
- Abbreviation: PA3N4MP
- Description: Red crystals
- 2-N-phenylamino-3-nitro-6-methylpyridine
- Chemical formula: C12H11N3O2
- Abbreviation: PA3N6MP
- Description: Red crystals
- Elemental chemical analysis:
- PA3N4MP (red crystals):
- ○
- Calculated: C 62.87%, H 4.84%, N 18.33%
- ○
- Experimental: C 62.51%, H 4.79%, N 18.17%
- ○
- Melting point: 395 K, 122 °C
- ○
- Synthetic yield: 79.0%
- PA3N6MP (red crystals):
- ○
- Calculated: C 62.87%, H 4.84%, N 18.33%
- ○
- Experimental: C 62.55%, H 4.88%, N 18.12%
- ○
- Melting point: 405 K, 132 °C
- ○
- Synthetic yield: 71.0%
3.2. Single-Crystal X-Ray Diffraction 128–142
3.3. Infrared and Raman Studies
3.4. Electronic Absorption Spectroscopy
3.5. Time-Resolved Emission Spectroscopy
3.6. Computational Methodology
3.7. NMR Spectroscopy
4. Conclusions
- In the present work, two new heterocyclic isomers, 2-N-phenylamino-4-methyl-3-nitro- and 2-N-phenylamino-6-methyl-3-nitro-pyridines, were obtained. Their structures were characterized on the basis of X-ray diffraction, IR and Raman spectra, as well as electron UV-Vis and emission spectra measurements. They form new class of pyridine derivatives with several potential applications. This follows from their peculiar structural, electronic, and optical properties.
- The specific structure of the studied compounds originates from the amino bridge between the phenyl and pyridine rings, and this conformation is stabilized by the intramolecular medium-strength hydrogen bond of N–H···O type formed by the O-atom of the nitro group and the NH group. The NH group plays an important role as the proton donor in the structural stabilization of the studied compounds in the solid state. Such structure predisposes these derivatives as precursors of antihistamine drugs, which can be used to oppose the activity of histamine receptors in the human body, forming antihistaminic pharmaceutics for allergic disorders. Such drugs contain the pyridine ring as an important part of their structure [68].
- Integrated spectroscopic analysis and DFT computational modeling reveal distinct singlet-triplet state distributions governing the photophysical behavior of the investigated pyridine isomers. Key findings include the following:
- ○
- Phosphorescence mechanism: electronic absorption and emission spectral data confirm that triplet→singlet phosphorescence arises predominantly through intersystem crossing machanism (ISC), a well-documented relaxation pathway in nitro-functionalized pyridine systems.
- ○
- Structure–function correlation: unique electronic architecture of phenyl-aminopyridine derivatives enables their dual functionality as follows:
- ■
- Versatile synthetic building blocks for heterocyclic chemistry;
- ■
- Efficient ligands for d-/f-block metal coordination complexes;
- ■
- Tunable luminophores with tailorable ISC efficiency.
- ○
- Technological potential: engineered lanthanide complexes incorporating these scaffolds demonstrate promise as follows:
- ■
- Targeted bioimaging probes leveraging metal-centered luminescence;
- ■
- Photosensitizers for photodynamic therapy applications;
- ■
- Molecular sensors for environmental metal ion detection.
- The 13C and 1H NMR spectra fully confirm the structural XRD data reported in the present work.
- The intense red color of the obtained materials allow for their use as new dyes for the production of the color plastic foils for food packing.
- This work establishes foundational structure–property relationships critical for rational design of next-generation luminescent materials in medicinal and materials chemistry.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Gazzar, A.-R.; Hussein, H.; Hafez, H. Synthesis and biological evaluation of thieno [2,3-d]pyrimidine derivatives for anti-inflammatory, analgesic and ulcerogenic activity. Acta Pharm. 2007, 57, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-H.; Savas, Ü.; Lasker, J.M.; Johnson, E.F. Genistein, Resveratrol, and 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside Induce Cytochrome P450 4F2 Expression through an AMP-Activated Protein Kinase-Dependent Pathway. J. Pharmacol. Exp. Ther. 2011, 337, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, A.M.R.; de Azevedo, A.R.; Pinheiro, L.C.d.S.; Borges, J.C.; Carvalho, V.L.; Miranda, M.D.; de Meneses, M.D.F.; Nascimento, M.; Ferreira, D.; Rebello, M.A.; et al. Synthesis and antiviral activity of new 4-(phenylamino)/4-[(methylpyridin-2-yl)amino]-1-phenyl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acids derivatives. Med. Chem. Res. 2007, 16, 352–369. [Google Scholar] [CrossRef]
- Wang, N.-Y.; Zuo, W.-Q.; Xu, Y.; Gao, C.; Zeng, X.-X.; Zhang, L.-D.; You, X.-Y.; Peng, C.-T.; Shen, Y.; Yang, S.-Y.; et al. Discovery and structure–activity relationships study of novel thieno[2,3-b]pyridine analogues as hepatitis C virus inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 1581–1588. [Google Scholar] [CrossRef]
- Elansary, A.K.; Moneer, A.A.; Kadry, H.H.; Gedawy, E.M. Synthesis and anticancer activity of some novel fused pyridine ring system. Arch. Pharmacal Res. 2012, 35, 1909–1917. [Google Scholar] [CrossRef]
- Lauria, A.; Abbate, I.; Patella, C.; Martorana, A.; Dattolo, G.; Almerico, A.M. New annelated thieno[2,3-e][1,2,3]triazolo[1,5-a]pyrimidines, with potent anticancer activity, designed through VLAK protocol. Eur. J. Med. Chem. 2013, 62, 416–424. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Al-Omran, F.; Azzam, R.A. Heterocyclic ring extension of estrone: Synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids 2014, 84, 46–56. [Google Scholar] [CrossRef]
- Nikkhoo, A.R.; Miri, R.; Arianpour, N.; Firuzi, O.; Ebadi, A.; Salarian, A.A. Cytotoxic activity assessment and c-Src tyrosine kinase docking simulation of thieno[2,3-b] pyridine-based derivatives. Med. Chem. Res. 2013, 23, 1225–1233. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Ahmed, O.M.; Elgendy, H.S. Novel synthesis of puriensanalougues and thieno [2, 3-b] pyridine derivatives with anticancer and antioxidant activity. J. Pharm. Res. 2014, 8, 1303–1313. [Google Scholar]
- Bakhite, E.A.; Abdel-Rahman, A.E.; Mohamed, O.S.; Thabet, E.A. Synthesis, reactions and antimicrobial activity of new cyclopenta [e] thieno [2, 3-b] pyridines and related heterocyclic systems. Pharmazie 2000, 55, 577–583. [Google Scholar] [CrossRef]
- Altalbawy, F.M.A. Synthesis and Antimicrobial Evaluation of Some Novel Bis-α,β-Unsaturated Ketones, Nicotinonitrile, 1,2-Dihydropyridine-3-carbonitrile, Fused Thieno [2,3-b]pyridine and Pyrazolo [3,4-b]pyridine Derivatives. Int. J. Mol. Sci. 2013, 14, 2967–2979. [Google Scholar] [CrossRef] [PubMed]
- Bumpus, N.N.; Johnson, E.F. 5-Aminoimidazole-4-carboxyamide-ribonucleoside (AICAR)-Stimulated Hepatic Expression of Cyp4a10, Cyp4a14, Cyp4a31, and Other Peroxisome Proliferator-Activated Receptor α-Responsive Mouse Genes Is AICAR 5′-Monophosphate-Dependent and AMP-Activated Protein Kinase-Independent. J. Pharmacol. Exp. Ther. 2011, 339, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Peyton, K.J.; Liu, X.-M.; Yu, Y.; Yates, B.; Durante, W. Activation of AMP-Activated Protein Kinase Inhibits the Proliferation of Human Endothelial Cells. J. Pharmacol. Exp. Ther. 2012, 342, 827–834. [Google Scholar] [CrossRef]
- Nakajima, K.; Komiyama, Y.; Hojo, H.; Ohba, S.; Yano, F.; Nishikawa, N.; Aburatani, H.; Takato, T.; Chung, U.-I. Enhancement of bone formation ex vivo and in vivo by a helioxanthin-derivative. Biochem. Biophys. Res. Commun. 2010, 395, 502–508. [Google Scholar] [CrossRef]
- Maeda, Y.; Hojo, H.; Shimohata, N.; Choi, S.; Yamamoto, K.; Takato, T.; Chung, U.-I.; Ohba, S. Bone healing by sterilizable calcium phosphate tetrapods eluting osteogenic molecules. Biomaterials 2013, 34, 5530–5537. [Google Scholar] [CrossRef]
- Gemba, T.; Ninomiya, M.; Matsunaga, K.; Ueda, M.J. Effects of a novel calcium antagonist, S-(+)-methyl-4, 7-dihydro-3-isobutyl-6-methyl-4-(3-nitrophenyl) thieno [2, 3-b] pyridine-5-carboxylate (S-312-d), on ischemic amino acid release and neuronal injury in stroke-prone spontaneously hypertensive rats. Pharmacol. Exp. Ther. 1993, 265, 463–467. [Google Scholar]
- Balzarini, J.; Stevens, M.; Andrei, G.; Snoeck, R.; Strunk, R.; Pierce, J.B.; Lacadie, J.A.; De Clercq, E.; Pannecouque, C. Pridine oxide derivatives: Structure-activity relationship for inhibition of human immunodeficiency virus and cytomegalovirus replication in cell culture. Helv. Chim. Acta 2002, 85, 2961–2974. [Google Scholar] [CrossRef]
- Stevens, M.; Pannecouque, C.; De Clercq, E.; Balzarini, J. Inhibition of human immunodeficiency virus by a new class of pyridine oxide derivatives. Antimicrob. Agents Chemother. 2003, 47, 2951–2957. [Google Scholar] [CrossRef]
- Stevens, M.; Pannecouque, C.; De Clercq, E.; Balzarini, J. Novel human immunodeficiency virus (HIV) inhibitors that have a dual mode of anti-HIV action. Antimicrob. Agents Chemother. 2003, 47, 3109–3116. [Google Scholar] [CrossRef]
- Balzarini, J.; Stevens, M.; De Clercq, E.; Schols, D.; Pannecouque, C. Pyridine N-oxide derivatives: Unusual anti-HIV compounds with multiple mechanisms of antiviral action. J. Antimicrob. Chemother. 2005, 55, 135–138. [Google Scholar] [CrossRef]
- Scriven, E.F.V.; Murugan, R. Pyridine and pyridine derivatives. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; Volume 20, pp. 1–53. [Google Scholar] [CrossRef]
- Liu, C.; Luo, J.; Xu, L.; Huo, Z. Synthesis of 2-substituted pyridines from pyridine N-oxides. Arkivoc 2013, 2013, 154–174. [Google Scholar] [CrossRef]
- Dzierszinski, F.; Coppin, A.; Mortuaire, M.; Dewailly, E.; Slomianny, C.; Ameisen, J.-C.; DeBels, F.; Tomavo, S. Ligands of the Peripheral Benzodiazepine Receptor Are Potent Inhibitors of Plasmodium falciparum and Toxoplasma gondii In Vitro. Antimicrob. Agents Chemother. 2002, 46, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Dressler, H. Pyridine and derivatives. In Van Nostrand’s Scientific Encyclopedia Van Nostrand’s Scientific Encyclopedia; Considine, D.M., Considine, G.D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Balzarini, J.; Keyaerts, E.; Vijgen, L.; Vandermeer, F.; Stevens, M.; De Clercq, E.; Egberink, H.; Van Ranst, M. Pyridine N-oxide derivatives are inhibitory to the human SARS and feline infectious peritonitis coronavirus in cell culture. J. Antimicrob. Chemother. 2005, 57, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. Arkivoc 2001, 2001, 242–268. [Google Scholar]
- Kilenyi, S.N.; Mousseau, J.J. Pyridine N-oxide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; p. 1. [Google Scholar] [CrossRef]
- Carlin, R.L. Transition Metal Complexes of Pyridine N-Oxide. J. Am. Chem. Soc. 1961, 83, 3773–3775. [Google Scholar] [CrossRef]
- Sarma, R.; Karmakar, A.; Baruah, J.B. Synthesis and characterization of pyridine N-oxide complexes of manganese, copper and zinc. Inorganica Chim. Acta 2008, 361, 2081–2086. [Google Scholar] [CrossRef]
- Lorenc, J. Dimeric structure and hydrogen bonds in 2-N-ethylamino-5-metyl-4-nitro-pyridine studied by XRD, IR and Raman methods and DFT calculations. Vib. Spectrosc. 2012, 61, 112–123. [Google Scholar] [CrossRef]
- Szatyłowicz, H. Structural aspects of the intermolecular hydrogen bond strength: H-bonded complexes of aniline, phenol and pyridine derivatives. J. Phys. Org. Chem. 2008, 21, 897–914. [Google Scholar] [CrossRef]
- Bryndal, I.; Kucharska, E.; Wandas, M.; Lorenc, J.; Hermanowicz, K.; Mączka, M.; Lis, T.; Marchewka, M.; Hanuza, J. Comprehensive physicochemical studies of a new hybrid material: 2-Amino-4-methyl-3-nitropyridinium hydrogen oxalate. Spectrochim. Acta A 2014, 117, 434–441. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 627–668. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld Surfaces: A New Tool for Visualising and Exploring Molecular Crystals. Chem. Eur. J. 1998, 4, 2136–2141. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 7, 3814–3816. [Google Scholar] [CrossRef]
- Ureña, F.; Gómez, M.; González, J.; Torres, E. A new insight into the vibrational analysis of pyridine. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 2815–2839. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Walters, V.A.; Wong, K.N.; Colson, S.D. Azines: Vibrational force field and intensities for pyridine. J. Phys. Chem. 1984, 88, 6067–6075. [Google Scholar] [CrossRef]
- Lorenc, J.; Bryndal, I.; Syska, W.; Wandas, M.; Marchewka, M.; Pietraszko, A.; Lis, T.; Mączka, M.; Hermanowicz, K.; Hanuza, J. Order–disorder phase transitions and their influence on the structure and vibrational properties of new hybrid material: 2-Amino-4-methyl-3-nitropyridinium trifluoroacetate. Chem. Phys. 2010, 374, 1–14. [Google Scholar] [CrossRef]
- Lorenc, J.; Hanuza, J.; Janczak, J. Structural and vibrational study of 3- or 5-methyl substituted 2-N-ethylamino-4-nitropyridine N-oxides. Vib. Spectrosc. 2012, 59, 59–70. [Google Scholar] [CrossRef]
- Bryndal, I.; Lorenc, J.; Macalik, L.; Michalski, J.; Sąsiadek, W.; Lis, T.; Hanuza, J. Crystal structure, vibrational and optic properties of 2-N-methylamino-3-methylpyridine N-oxide–Its X-ray and spectroscopic studies as well as DFT quantum chemical calculations. J. Mol. Struct. 2019, 1195, 208–219. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Bryndal, I.; Kucharska, E.; Sąsiadek, W.; Wandas, M.; Lis, T.; Lorenc, J.; Hanuza, J. Molecular and crystal structures, vibrational studies and quantum chemical calculations of 3 and 5-nitroderivatives of 2-amino-4-methylpyridine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 96, 952–962. [Google Scholar] [CrossRef]
- Bryndal, I.; Marchewka, M.; Wandas, M.; Sąsiadek, W.; Lorenc, J.; Lis, T.; Dymińska, L.; Kucharska, E.; Hanuza, J. The role of hydrogen bonds in the crystals of 2-amino-4-methyl-5-nitropyridinium trifluoroacetate monohydrate and 4-hydroxybenzenesulfonate–X-ray and spectroscopic studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 123, 342–351. [Google Scholar] [CrossRef]
- Lorenc, J.; Kucharska, E.; Hanuza, J.; Chojnacki, H. Excited electronic states of 2-ethylamino-(3 or 5)-methyl-4-nitropyridine and 2-methylamino-(3 or 5)-methyl-4-nitropyridine. J. Mol. Struct. 2004, 707, 47–57. [Google Scholar] [CrossRef]
- Lorenc, J.; Zając, A.; Janczak, J.; Lisiecki, R.; Hanuza, J.; Hermanowicz, K. Structure and optical properties of new nitro-derivatives of 2-N-alkiloamino-picoline N-oxide isomers. J. Mol. Struct. 2022, 1265, 133372. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Najjar, H.J.; Al-Majid, A.M.; Soliman, S.M.; Mabkhot, Y.N.; Shaik, M.R.; Ghabbour, H.A.; Fun, H.-K. Synthesis, NMR, FT-IR, X-ray structural characterization, DFT analysis and isomerism aspects of 5-(2,6-dichlorobenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 147, 107–116. [Google Scholar] [CrossRef]
- Carthigayan, K.; Xavier, S.; Periandy, S. HOMO–LUMO, UV, NLO, NMR and vibrational analysis of 3-methyl-1-phenylpyrazole using FT-IR, FT-RAMAN FT-NMR spectra and HF-DFT computational methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 142, 350–363. [Google Scholar] [CrossRef]
- Puszko, A.; Wasylina, L. The influence of steric effect on 1H NMR, 13C NMR and IR spectra of methylated de-rivatives of 4-nitropyridine N-oxide. Chem. Pap. 1995, 49, 176–181. [Google Scholar]
- Kątcka, M.; Urbański, T. NMR spectra of pyridine, picolines and hydrochlorides and of their hydrochlorides and methiodides. Bull. Acad. Pol. Sci. 1968, 16, 347–350. [Google Scholar]
- Palasek, B.; Talik, Z. Synthesis of the 2-alcilonitramino-5-nitroderivatives of 6-methylpyridine. Pr. Nauk. AE Wrocław 1985, 291, 111–114. [Google Scholar]
- Rigaku Oxford Diffraction, CrysAlisPro 1.171.42.93a Software System, Oxford, UK. 2023. Available online: https://rigaku.com/products/crystallography/x-ray-diffraction/crysalispro (accessed on 28 February 2025).
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure of Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND, Version 3.0; Crystal Impact GbR: Bonn, Germany, 2006.
- Wolf, S.W.; Grimwood, D.J.; MacKimon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, A.M. Crystal Explorer, Ver. 3.1; University of Western Australia: Perth, Australia, 2013.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision A.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, B37, 785–789. [Google Scholar]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Rostkowska, H.; Lapinski, L.; Nowak, M.J. Analysis of the normal modes of molecules with D3h symmetry: Infrared spectra of monomeric s-triazine and cyanuric acid. Vib. Spectrosc. 2009, 49, 43–51. [Google Scholar] [CrossRef]
- GaussView 4.1; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Zhurko, G.A.; Zhurko, D.A. Chemcraft Graphical Program for Visualization of Computed Results. Available online: http://www.chemcraftprog.com (accessed on 10 August 2024).
- Michalska, D. RAINT (Raman Intensities), Computer Program for Calculation of Raman Intensities from the Gaussian Outputs; Wrocław University of Technology: Wrocław, Poland, 2002. [Google Scholar]
- Palafox, M.A.; Rastogi, V. Quantum chemical predictions of the vibrational spectra of polyatomic molecules. The uracil molecule and two derivatives. Spectrochim. Acta 2002, A58, 411–440. [Google Scholar] [CrossRef]
- Joshi, K.K. Chemistry with Schiff bases of pyridine derivatives: Their potential as bioactive ligands and chemosensors. In The Edited Volume: Exploring Chemistry with Pyridine Derivatives; Pal, S., Ed.; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X-Ray | DFT | ||||||
|---|---|---|---|---|---|---|---|
| N3–O1 | 1.2412(15) | 1.242 | |||||
| N3–O2 | 1.2272 (14) | 1.226 | |||||
| C3–N3 | 1.4508 (16) | 1.454 | |||||
| C2–N2–C7 | 131.01 (11) | 131.64 | |||||
| Dihedral angle between the planes: | |||||||
| NO2 (O1N3O2)/pyridine ring (N1,C2–C6) | 3.84(15) | 25.50 | |||||
| pyridine ring (N1,C2–C6)/phenyl ring (C7–C12) | 6.20(15) | 15.70 | |||||
| D–H···A | D–H | H···A | D···A | D–H···A | |||
| N2–H2···O1 | 0.900(16) | 1.860(15) | 2.5877(14) | 136.4(13) | X-ray | ||
| N2–H2···O1 | 1.014 | 1.814 | 2.632 | 135.17 | DFT | ||
| X-Ray | DFT | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N3–O1 | 1.249 (2) | 1.243 | ||||||||
| N3–O2 | 1.230 (2) | 1.226 | ||||||||
| C3–N3 | 1.447 (3) | 1.450 | ||||||||
| C2–N2–C7 | 132.0 (2) | 132.24 | ||||||||
| Dihedral angle between the planes: | ||||||||||
| NO2 (O1N3O2)/pyridine ring (N1,C2–C6) | 3.84 (15) | 0.00 | ||||||||
| pyridine ring (N1,C2–C6)/phenyl ring (C7–C12) | 6.20 (15) | 0.00 | ||||||||
| D–H···A | D–H | H···A | D···A | D–H···A | ||||||
| N2–H2···O1 | 0.84 (2) | 1.96 (2) | 2.649 (3) | 139 (2) | X-ray | |||||
| N2–H2···O1 | 1.016 | 1.816 | 2.649 | 136.58 | DFT | |||||
| PA3N4MP | PA3N6MP | |||||||
|---|---|---|---|---|---|---|---|---|
| Calc. | Exp. | Calc. | Exp. | |||||
| IR | RS | IR | RS | IR | RS | IR | RS | Assignment |
| 3367 | 3367 | 3433m | 3344 | 3344 | 3311w | νN–H····O | ||
| 1585 | 1585 | 1579s | 1579w | 1589 | 1589 | 1580vs | 1585w | δ(CNH)ϕ + ν(ϕ) |
| 1574 | 1574 | 1566s | 1567w | 1578 | 1578 | 1578w | δ(CNH)θ + ν(ϕ) + νas(NO2) | |
| 1549 | 1549 | 1548s | 1547m | 1548 | 1548 | 1548vw | 1545w | νas(NO2) + ν(ϕ) + ν(θ) + δ(Cϕ NCθ) |
| 1350 | 1350 | 1361 | 1357 | 1357 | 1357w | ν(CN)ϕ + νs(NO2) + δ(ϕ) + δs(CH3) | ||
| 1226 | 1226 | 1237m | 1212s | 1227 | 1227 | 1219vs | 1216s | ν(ϕ) + νs(NO) + ν(CN)ϕα + ν(CNH)ϕ |
| 1197 | 1197 | 1220s | 1192sh | 1195 | 1195 | 1186s | 1188s | νs(NO2) + (CNH)θ |
| 1184 | 1184 | 1203sh | νs(NO2) + ν(NCN)ϕ | |||||
| 807 | 807 | 807m | ν(CNHC) | |||||
| 790 | 790 | ν(CNHC)ϕ+θ + δs(NO)ϕ | ||||||
| 707 | 707 | 710 | 710 | δ(NH) | ||||
| 699 | 699 | 691m | 694m | δ(CNHC)ϕ+θ | ||||
| 395 | 395 | 386w | 386 | 386 | 394w | 395vw | δ(CCH3) + δ(CNO)ϕ | |
| 290 | 290 | 289w | 295m | 295vw | δ(CNHC)ϕ+θ | |||
| 187 | 187 | 184 | 184 | 188w | δ(CNHC)ϕ+θ | |||
| 107 | 107 | 94 | 94 | 177w | δ(CNHC)ϕ+θ | |||
| 83 | 83 | 84 | 84 | 150m | δs(NO)ϕ + δ(CNHC)ϕ+θ | |||
| (a) | ||||
| Electron levels | eV | nm | cm−1 | Oscillator strength |
| singlets | ||||
| (1) | 2.6717 | 464.06 | 21,552 | 0.1230 |
| (2) | 2.7783 | 446.25 | 22,409 | 0.1648 |
| (3) | 3.6225 | 342.26 | 29,218 | 0.0008 |
| (4) | 3.6351 | 341.07 | 29,319 | 0.0009 |
| (5) | 3.7309 | 332.32 | 30,092 | 0.0022 |
| (6) | 3.7385 | 331.64 | 30,153 | 0.0026 |
| (7) | 4.0091 | 309.26 | 32,336 | 0.0038 |
| (8) | 4.1820 | 296.47 | 33,730 | 0.1643 |
| (9) | 4.2399 | 292.42 | 34,197 | 0.1730 |
| (10) | 4.2476 | 291.89 | 34,260 | 0.0935 |
| (11) | 4.3350 | 286.00 | 34,965 | 0.1234 |
| (12) | 4.5328 | 273.53 | 36,559 | 0.0007 |
| (13) | 4.6380 | 267.32 | 37,408 | 0.1533 |
| (14) | 4.8088 | 257.83 | 38,785 | 0.0237 |
| (15) | 4.9383 | 251.07 | 39,830 | 0.2944 |
| (16) | 5.0938 | 243.40 | 41,085 | 0.3909 |
| (17) | 5.2701 | 235.26 | 42,506 | 0.0019 |
| (18) | 5.3092 | 233.53 | 42,821 | 0.0295 |
| (19) | 5.5492 | 223.43 | 44,757 | 0.0007 |
| (20) | 5.8961 | 210.28 | 47,556 | 0.0212 |
| triplets | ||||
| (1) | 1.6711 | 741.93 | 13,478 | 0.0000 |
| (2) | 1.7361 | 714.16 | 14,002 | 0.0000 |
| (3) | 1.8193 | 681.49 | 14,674 | 0.0000 |
| (4) | 1.9895 | 623.19 | 16,046 | 0.0000 |
| (5) | 2.1489 | 576.96 | 17,332 | 0.0000 |
| (6) | 2.7649 | 448.42 | 22,300 | 0.0000 |
| (b) | ||||
| Electron levels | eV | nm | cm−1 | Oscillator strength |
| singlets | ||||
| (1) | 2.7315 | 453.91 | 22,031 | 0.1285 |
| (2) | 2.8418 | 436.28 | 22,921 | 0.1693 |
| (3) | 3.7076 | 334.40 | 29,904 | 0.0000 |
| (4) | 3.7191 | 333.37 | 29,997 | 0.0001 |
| (5) | 3.7537 | 330.30 | 30,030 | 0.0007 |
| (6) | 3.7585 | 329.88 | 30,314 | 0.0010 |
| (7) | 3.9573 | 313.31 | 31,917 | 0.0000 |
| (8) | 3.9688 | 312.40 | 32,010 | 0.0001 |
| (9) | 4.2224 | 293.63 | 34,056 | 0.2176 |
| (10) | 4.3072 | 287.86 | 34,739 | 0.2413 |
| (11) | 4.5014 | 275.43 | 36,307 | 0.0007 |
| (12) | 4.6978 | 263.92 | 37,890 | 0.0396 |
| (13) | 4.9311 | 251.43 | 39,773 | 0.0334 |
| (14) | 5.0652 | 244.78 | 40,853 | 0.2846 |
| (15) | 5.2259 | 237.25 | 42,150 | 0.4088 |
| (16) | 5.3044 | 233.74 | 42,783 | 0.0018 |
| (17) | 5.5145 | 224.83 | 44,478 | 0.0449 |
| (18) | 5.6807 | 218.25 | 45,819 | 0.0095 |
| (19) | 5.8055 | 213.56 | 46,825 | 0.0101 |
| (20) | 5.9308 | 209.05 | 47,835 | 0.0233 |
| triplets | ||||
| (1) | 1.6560 | 748.71 | 13,356 | 0.0000 |
| (2) | 1.7222 | 719.90 | 13,891 | 0.0000 |
| (3) | 1.7516 | 707.82 | 14,128 | 0.0000 |
| (4) | 1.9444 | 637.65 | 15,683 | 0.0000 |
| (5) | 2.0811 | 595.77 | 16,785 | 0.0000 |
| (6) | 2.2669 | 546.94 | 18,284 | 0.0000 |
| (7) | 2.5069 | 494.58 | 20,219 | 0.0000 |
| (8) | 2.5736 | 481.76 | 20,757 | 0.0000 |
| (9) | 2.8381 | 436.86 | 22,891 | 0.0000 |
| (10) | 3.0912 | 401.08 | 24,933 | 0.0000 |
| PA3N4MP | PA3N6MP | |
|---|---|---|
| Formula | C12H11N3O2 | C12H11N3O2 |
| Mol. weight | 229.24 | 229.24 |
| Temperature (K) | 100(2) | 100(2) |
| Crystal system | triclinic | monoclinic |
| Crystal color | red | red |
| Space group | P-1 | P21/n |
| a (Å) | 7.3285(5) | 7.8874(5) |
| b (Å) | 7.4403(6) | 17.3681(11) |
| c (Å) | 10.9340(11) | 8.4396(7) |
| α (°) | 101.781(8) | |
| β (°) | 102.348(7) | 111.309(9) |
| γ (°) | 109.981(7) | |
| V (Å3) | 521.79(8) | 1077.09(15) |
| Z | 2 | 4 |
| Dcal | 1.459 | 1.414 |
| θ range (°) | 3.056–29.312 | 2.844–29.331 |
| μ (mm−1) | 0.103 | 0.100 |
| Crystal size | 0.32 × 0.24 × 0.13 | 0.27 × 0.09 × 0.08 |
| Tmin./Tmax. | 0.9825/1.000 | 0.9792/1.000 |
| Total/unique/obs. refls. | 4590/2420/1955 | 7173/2565/1515 |
| Rint | 0.0158 | 0.0545 |
| R [F2 > 2σ(F2)] a | 0.0414 | 0.0650 |
| wR [F2 all refls] a | 0.1129 | 0.1359 |
| S | 0.978 | 1.001 |
| Δρmax, Δρmin (eÅ−3) | +0.228; −0.324 | +0.244; −0.247 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godlewska, P.; Hanuza, J.; Janczak, J.; Lisiecki, R.; Basiak, M.; Zając, A.; Dymińska, L. Structure and Optical Properties of New 2-N-Phenylamino-methyl-nitro-pyridine Isomers. Int. J. Mol. Sci. 2025, 26, 2874. https://doi.org/10.3390/ijms26072874
Godlewska P, Hanuza J, Janczak J, Lisiecki R, Basiak M, Zając A, Dymińska L. Structure and Optical Properties of New 2-N-Phenylamino-methyl-nitro-pyridine Isomers. International Journal of Molecular Sciences. 2025; 26(7):2874. https://doi.org/10.3390/ijms26072874
Chicago/Turabian StyleGodlewska, Patrycja, Jerzy Hanuza, Jan Janczak, Radosław Lisiecki, Małgorzata Basiak, Adam Zając, and Lucyna Dymińska. 2025. "Structure and Optical Properties of New 2-N-Phenylamino-methyl-nitro-pyridine Isomers" International Journal of Molecular Sciences 26, no. 7: 2874. https://doi.org/10.3390/ijms26072874
APA StyleGodlewska, P., Hanuza, J., Janczak, J., Lisiecki, R., Basiak, M., Zając, A., & Dymińska, L. (2025). Structure and Optical Properties of New 2-N-Phenylamino-methyl-nitro-pyridine Isomers. International Journal of Molecular Sciences, 26(7), 2874. https://doi.org/10.3390/ijms26072874