Abstract
Gliomas are the most prevalent malignant tumors in the adult central nervous system (CNS). Glioblastoma (GBM) accounts for approximately 60–70% of primary gliomas. It is a great challenge to human health because of its high degree of malignancy, rapid progression, short survival time, and susceptibility to recurrence. Owing to the specificity of the CNS, the glioma microenvironment often contains numerous glial cells. Astrocytes are most widely distributed in the human brain and form reactive astrocyte proliferation regions around glioma tissue. In addition, astrocytes are activated under pathological conditions and regulate tumor and microenvironmental cells through cell-to-cell contact or the secretion of active substances. Therefore, astrocytes have attracted attention as important components of the glioma microenvironment. Here, we focus on the mechanisms of reactive astrocyte activation under glioma conditions, their contribution to the mechanisms of glioma genesis and progression, and their potential value as targets for clinical intervention in gliomas.
1. Introduction
The latest epidemiological reports indicate that the overall incidence of malignant brain tumors in adults is approximately 7 per 100,000 population, with an overall 5-year survival rate of 36% [1]. The overall percentage of gliomas is about 80-85%, with glioblastoma (GBM) accounting for 49% and diffuse lower-grade glioma (LGG) accounting for 30% among malignant brain tumors [2]. Currently, the most used clinical management paradigms for gliomas are surgery combined with radiotherapy and temozolomide chemotherapy. In the case of GBM, the 2-year and 5-year survival rates for combination therapy are 27.2% and 9.8%, respectively, significantly improved compared with 10.9% and 1.9% for monotherapy [1]. Even so, gliomas remain difficult to cure and are one of the great threats to human life and health.
As one of the fundamental hallmarks of cancer, the formation of the tumor microenvironment significantly contributes to the development and progression of gliomas [3,4]. In the early years, studies related to the glioma microenvironment were mainly focused on immune cells in the traditional sense (e.g., T and B cells, monocytes, dendritic cells, etc.) and microglia. As for astrocytes, they are widely present in the glioma microenvironment, and pathologists have found areas of astrocyte proliferation around glioma tissues. Since gliomas also contain tumors of astrocyte origin (also known as astrocytomas), the early focus of scientists tended to be on how to differentiate between tumorigenic and nontumorigenic astrocytes or to explore the mechanisms by which astrocytes malignantly transform into glioma cells [5,6,7]. However, this has also led to a relative underappreciation of the biological significance of astrocytes as members of the microenvironment in glioma genesis, progression, and evolution.
Reviewing the research data on reactive astrocytes, we see that their development has proceeded through several major stages. The origins of this scientific problem lie in a phenomenon that is often accompanied by lesions of the central nervous system (CNS) called “reactive astrogliosis”. As neuroscience progressed, scientists noticed that reactive astrocyte “scarring” was also present in a variety of neuroimmune and degenerative diseases such as multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease. As a result, the neurotrophic and supportive capacity of astrocytes has been implicated in the neuronal degeneration and deficits of these diseases, and abnormal reactive astrocytes in pathological states are thought to be neurotoxic. For example, referring to the macrophage/microglia M1/M2 typing strategy, reactive astrocytes can also be roughly classified into two phenotypes named A1 and A2 [8,9], with the A1 phenotype considered neurotoxic and the A2 phenotype having neuroprotective functions such as the promotion of axonal injury repair and neuronal regeneration [8,10]. Notably, the model of pathology induction in A1 relies on cytokines such as TNF-α, IL-1α, and C1q secreted by microglia after lipopolysaccharide stimulation [8]. Also, multiple complement cascade regulators are upregulated in reactive astrocytes in A1. They can exert a wide range of regulatory effects on immune and non-immune cells via fibroblast growth factor (FGF), C-X-C Motif Chemokine Ligand 1 (CXCL1), C-X-C Motif Chemokine Ligand 2 (CXCL2), Interleukin 6 (IL6), etc. [8,10,11,12]. Conversely, A2 can inhibit the immune response by releasing TGF-β, protecting the CNS from damage due to excessive inflammatory responses [12]. Thus, astrocytes are essential regulators of inflammation in the CNS. Notably, immune escape stands as one of the defining hallmarks of gliomas. Some characteristic molecules of certain reactive astrocyte subsets have been widely investigated within the glioma context. For example, TGF-β plays a pivotal role in sustaining glioma proliferation, invasion, and immune evasion. It can achieve this by preserving the viability of glioma stem cells (GSCs), recruiting Tumor-Associated Macrophages (TAMs), and inducing the exhaustion of CD8+ T cells and NK cells [13,14,15,16]. This has inspired oncologists to consider whether reactive astrocytes in the glioma microenvironment are merely “candidates” for carcinogenesis or whether they can act as organizers and participants in the formation and evolution of the glioma microenvironment.
Encouragingly, there is now a fundamental understanding that glioma-reactive astrocytes are widespread in the microenvironment and can exert pro-tumorigenic effects. Astrocytes activated by stimuli from glioma cells, microglia, and hypoxic conditions can enhance the proliferative, invasive, and metastatic capabilities of glioma cells through the secretion of effector molecules or via direct contact facilitated by gap junctions [17,18,19,20,21,22,23,24,25]. In organotypic slice models and in vivo mouse and rat models, the depletion of astrocytes has been significantly observed to cause a reduction in the in situ growth rates of gliomas [26,27,28]. Moreover, reactive astrocytes are linked to the generation of resistance to radiation and chemotherapy in gliomas [29,30]. They also correlate with the recurrence of gliomas after surgical resection [26,31]. Consequently, intervention strategies targeting reactive astrocytes hold promise as a potential adjuvant approach for the treatment of gliomas.
Here, we focus on the mechanisms of interaction between reactive astrocytes and microglia, circulating immune cells, and tumor cells in the context of glioma after having been validated in cellular or zoological models. At the same time, we focus on the mechanisms and heterogeneity of astrocytes acquiring reactivity in the glioma microenvironment and their potential value during clinical interventions in gliomas. Figure 1 briefly summarizes the acquisition of reactivity by astrocytes in the glioma microenvironment.
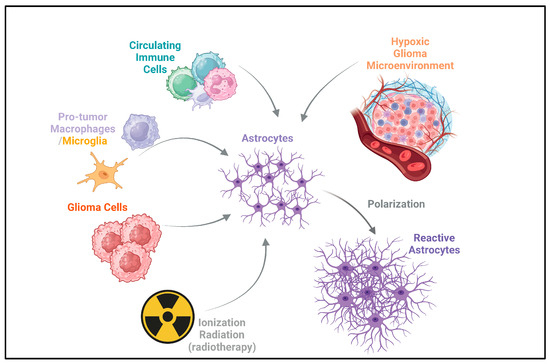
Figure 1.
Acquisition of reactivity by astrocytes in glioma. Astrocytes can acquire reactivity through multiple ways, including circulating immune cells, pro-tumor macrophages/microglia, and glioma cells. Ionizing radiation from radiotherapy also acts on astrocytes [17,18,19,20,21,22,23,24,25,26,27,28]. This can potentially induce reactivity in astrocytes, which may contribute to the recurrence of gliomas after radiotherapy [30,32]. Additionally, the hypoxic glioma microenvironment drives the polarization of astrocytes, leading to the formation of reactive astrocytes [24]. The arrows depict the direction of influence and interaction among these components in the glioma microenvironment.
2. Astrocytes and Glioma Oncogenesis
There are three major viewpoints regarding glioma oncogenesis: the hypothesis of glioma stem cell origin, the hypothesis of neural stem cell origin, and the hypothesis of glial cell origin. The proposal, development, and refinement of these hypotheses largely rely on the development of single-cell RNA sequencing (scRNA-seq) technology and the advanced algorithms derived from it [33,34]. This enables us to further search for, subdivide, and define the sub-populations of glioma cells, as well as, to some extent, to infer their origins and directions of differentiation. Glioblastoma cells (WHO 4, IDH-wt) can be classified into four classic states based on scRNA-seq profiles and malignancy: oligodendrocyte progenitor cell-like (OPC-like), astrocyte-like (AC-like), neuronal progenitor cell-like (NPC-like), and mesenchymal-like (MES-like) subtypes [34]. In these subtypes, AC-like glioma cells express some characteristic astrocyte markers, such as S100B, GFAP, SLC1A3, GLAST, and MLC1. As is well known, tumor phenotypic transformation results from stage-specific transcriptional changes in progenitor cells. Currently, there is no clear definition of astrocyte precursors. However, under specific conditions, such as traumatic brain injury, astrocytes can redifferentiate and acquire the ability to enter the cell cycle for proliferation [35]. In addition, Li et al. mentioned in a cross-lineage analysis study that mouse cortical radial glial cells can give rise to ASCL1⁺EGFR⁺ apical multipotent intermediate progenitor cells, which then differentiate into basal multipotent intermediate progenitor cells expressing achaete-scute complex-like 1 (ASCL1), epidermal growth factor receptor (EGFR), oligodendrocyte transcription factor 2 (OLIG2), and proliferating cell nuclear antigen Ki-67 (MKI67). These progenitor cells can later transform into astrocyte lineage-restricted progenitor cells [36]. This evidence indicates that astrocyte precursors may persist in the CNS and can differentiate to replenish astrocytes. Astrocytes may also redifferentiate under specific conditions, enhancing their proliferative capacity. These processes can be regarded as the event nodes of astrocytes. The oncogene and tumor suppressor gene mutations occurring therein may lead to glioma tumorigenesis. For example, in in vitro models, the in situ injection of astrocytes with oncogene and suppressor gene mutations can lead to the development of glioma. At the same time, the glioma cells formed in this way all express common astrocyte markers, such as glial fibrillary acidic protein (GFAP) [37,38,39]. Inducing the combined deletion of phosphatase and tensin homolog (PTEN), tumor protein p53 (TP53), and retinoblastoma 1 (RB1) in mouse astrocytes can lead to the malignant progression of glioma, manifested as a transition from WHO grade III to WHO grade IV [40]. These findings support the astrocyte origin part of the hypothesis of glial cell origin. However, it is too one-sided or incomplete to explain the origin of glioma with a single hypothesis. First, the hypothesis of glial cell origin cannot account for the origin of some glioma cell subsets, such as NPC-like cells. These cells characteristically express neural progenitor cell markers, such as SOX4, SOX11, and DCX [34]. Neural stem cells/progenitor cells are the starting point for the development of glial cells and nerve cells. Whether glioma cells originating from astrocytes can redifferentiate and form a phenotype like that of neural progenitor cells remains debatable. Furthermore, since neural progenitor cells can differentiate into the astrocyte lineage, does it mean that tumor cells with an astrocyte-like phenotype can originate from the downward differentiation of NPC-like cells? Secondly, GSCs, like NPC-like cells, also widely exist in glioma tissues of various pathological types and grades. They characteristically express stem cell factors such as CD133, sex-determining region Y-box 2 (SOX2), Nestin, and OCT-4 [41]. These cells possess the abilities of self-renewal, differentiation, and resistance to DNA damage, and are important drivers of glioma progression [42,43]. Unfortunately, the glial cell origin hypothesis also fails to adequately explain the generation and formation of GSCs.
In summary, we tend to believe that the oncogenesis of gliomas may occur in the form of multiple cell sources, and glioma heterogeneity may not only result from the mutations or differentiation of a single cell line. The formation of glioma is a process of systemic breakdown in the CNS. The malignant transformation of astrocytes may be a crucial part of this process, or it could be the result of the accumulation of risk factors after the formation of basic glioma tissues. Elucidating these questions is significant for a clearer and deeper understanding of the glioma oncogenesis process.
Besides serving as a potential source of malignant cells, astrocytes have a robust ability to synthesize and secrete substances. This implies that non-malignant astrocytes can work together with glioma cells, microglia, macrophages, circulating immune cells, etc., to establish the glioma microenvironment and aid in the malignant progression of glioma [24,44,45,46]. These non-malignant astrocytes generally acquire reactivity upon receiving stimuli such as hypoxia and inflammatory factors in the microenvironment. Subsequently, we refer to them as “glioma-reactive astrocytes”.
3. Identification of Glioma-Reactive Astrocytes
Currently, the definition of glioma-reactive astrocytes is mainly based on findings in inflammatory diseases of the CNS. Common biomarkers include GFAP, s100 calcium-binding protein B (S100B), s100 calcium-binding protein A10 (S100A10), complement component 1 q subcomponent (C1q), neuroepithelial stem cell protein (NES), vimentin (VIM), and complement component 3 (C3). As early as 1978, astrocytes were shown to be more common in glioma tissues with relatively normal cell morphology typically distinct from malignant cells [47]. However, unlike the astrocytes in normal brain tissue, their volume is usually increased together with more branches. Although the concept of reactive astrocytes was lacking during that period, the authors described them as “well-preserved astrocytes”. Secondly, in an experimental model study, GFAP expression was also upregulated in astrocytes co-cultured with U87 [48]. GFAP is a crucial constituent of the astrocyte cytoskeleton. In the context of astrocytes undergoing reactive transformation, a conspicuous increase in both cell volume and the number of cell branches occurs. This reactive process is invariably accompanied by the upregulation of GFAP expression [49]. Thus, secretions from glioma cells can contribute to the acquisition of reactivity by astrocytes. Meanwhile, transforming growth factor-β (TGF-β), acid secretory protein (SPARC), and matrix metallopeptidase 2 (MMP-2) protein levels were also significantly elevated in U87 co-cultured astrocytes [48]. This further substantiates the existence of glioma-reactive astrocytes and indicates that they may be involved in the regulation of glioma immunity and the formation of an immunosuppressive microenvironment. S100B is an EF-hand-type Ca (2+)-binding protein highly expressed in astrocytes, which has various functions such as regulating Ca (2+)-homeostasis in the CNS and modulating cell proliferation and differentiation. Under pathological conditions, the S100B protein can regulate inflammatory responses through the MAPK and NF-κB pathways and mediate neurological damage by promoting oxidative stress [50,51]. In the context of gliomas, S100B can stimulate the phosphatidylinositol 3 -kinase/ protein kinase B (PI3K/Akt) and PI3K/RhoA pathways by interacting with Rous sarcoma virus-related tyrosine-protein kinase src (Src kinase), which is involved in the activation of astrocytes [52]. S100B can also maintain immunosuppressive microenvironmental characteristics of gliomas by chemotaxis of TAMs through the upregulation of CCL2 [53]. Notably, changes in the expression of markers such as S100A10, C1q, NES, VIM, and C3 and their mechanisms of regulating reactivity have only been investigated in reactive astrocytes under non-tumor disease conditions [8,10,12]. In current experimental studies, the identification of glioma-reactive astrocytes also refers to the differential expression patterns of these markers. However, due to the specificity of gliomas, reactive astrocytes may be different in the context of gliomas than in general CNS inflammatory diseases. The activation mechanisms and the consequent effects of the expression of these markers in glioma-reactive astrocytes, along with the question of whether there exist more specific markers for glioma-reactive astrocytes, are all issues that merit further in-depth investigation.
The advancement and popularization of sequencing technologies (including bulk-/sc-/sn-RNA-seq, etc.) have provided new insights to address this issue. Al-Dalahmah et al. characterized the microenvironment of GBM using snRNA-seq and clustered GFAP-positive reactive astrocytes into three subtypes: protoplasmic astrocytes (AST1, highly expressing SLC1A2/3/4, CPE, CPG5), reactive astrocytes expressing oligodendrocyte and neuronal genes (AST2, high expression of PLP1, RPL13A, RPL31, FTL, BCYRN1), and reactive astrocytes expressing inflammatory genes (AST3, high expression of S100B, CP, C3, CD44, CHI3L1/2, CLU) [54]. By comparing with CNV-pos tumor cells, synaptic nucleoprotein genes (SNCA, SNCB, and SNCG), WIF1, CHI3L2, ALDOC, ALDOA, AQP4, carbonic anhydrases CA2 and CA11, and CXCL14 were characteristically highly expressed in reactive astrocytes. Henrik Heiland et al. performed a linear downscaling of astrocyte subpopulations in the GBM microenvironment after scRNA-seq sequencing based on the “Partitioning Around Medoids” algorithm. Six reactive astrocyte subtypes at different stages of differentiation were identified by gene set enrichment analysis [28]. Compared to simply using generic markers such as GFAP, S100B, and VIM for the identification of reactive astrocytes, this approach is currently relatively more concerned with the pathophysiological functions of the cells. This helps us to better understand and discover the functional localization of different reactive astrocyte subpopulations in the glioma microenvironment and the mechanisms behind their pro-tumorigenic effects. However, due to the limited sample size, the generalizability of these typing strategies is not yet known, and the academic community has not reached a consensus on this issue. Mechanisms of reactive astrocyte heterogeneity generation in the glioma microenvironment remain highly explorable in the future.
4. Interaction Between Reactive Astrocytes and Glioma Cells
Reactive astrocytes regulate the malignant progression of gliomas through several complex mechanisms. Most directly, reactive astrocytes can interact with tumor cells, thereby promoting their proliferation, invasion, and migration. This capacity depends on direct cell-to-cell contact mediated by membrane proteins on the surface of astrocytes or molecules delivered by non-contact means such as paracrine and extracellular vesicles. Secondly, normal informational interactions between astrocytes and other CNS components may be disrupted by gliomas, thereby facilitating their development and progression. Based on the available evidence, we initiated the review of this issue.
4.1. Abnormalities in Direct Cell-to-Cell Contact
Under physiological conditions, astrocytes have important CNS homeostatic maintenance functions, such as providing energy and nutrient support to neurons, balancing intra- and extracellular ionic and water homeostasis, scavenging neurotransmitters from synapses, participating in the formation and modulation of the blood–brain barrier, and regulating neuronal and synaptic plasticity based on gliotransmitter release [55,56]. These functions cannot be separated from the widespread physical connections between astrocytes or neurons/synapses, with gap junctions (GJs) dominated by the channel junction proteins CX43 and CX30 [57,58,59]. Pathologic changes in the CNS are often accompanied by abnormalities in the function of the material information exchange channels formed by the GJs. For example, during the formation of epileptic foci, disruption of GJ coupling in the astrocyte network leads to a reduction in the buffering capacity of potassium ions and the accumulation of intracellular sodium ions. Excess extracellular potassium ions contribute to the acquisition of abnormal excitability of neurons, and the accumulation of intracellular sodium ions in astrocytes further impairs the clearance of glutamate within the synapse, exacerbating the abnormal excitability of neurons [60,61]. Interestingly, altered membrane potential in cancer cells appear to be associated with glioma progression. For example, Venkatesh HS et al. discovered that non-synapse-dependent potassium currents can depolarize glioma membranes and promote their proliferation [62]. Notably, epilepsy is a common clinical manifestation of gliomas. In terms of pathogenesis, both glioma and epilepsy have been reported to be accompanied by the disorder of calcium ion regulation in astrocytes [63]. Thus, gliomagenesis might be potentially related to the disruption of astrocyte functional networks and extracellular ionic homeostasis. However, the relationship between homeostatic dysregulation associated with disruption of astrocyte functional networks and the development and progression of gliomas is still not fully elucidated. Current studies tend to be more inclined to focus on the abnormalities of glioma tissue but unconsciously ignore the subsequent effects caused by the disruption of normal CNS homeostasis. This issue deserves further exploration in the future.
Secondly, direct biphasic interactions of information and material between glioma cells and astrocytes can also be realized through GJs (Figure 2). Since 1999, glioma cells and astrocytes have been shown to produce physical connections via CX43 under co-culture conditions. Also, the morphology of the corresponding astrocytes is significantly altered, as evidenced by an increase in cell volume and the number of membrane protrusions [64]. Sin et al. found that the knockdown of CX43 in astrocytes resulted in a decrease in the migration of cancer cells from the core to the periphery of mouse glioma models, demonstrating that CX43-mediated reactive astrocyte-tumor cell signaling might be important in regulating the invasion and migration of gliomas [19]. Further studies have indicated that the second messenger cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) can translocate from carcinoma cells into astrocytes via CX43-mediated cell–cell junctions, activate the STING pathway to increase their reactivity, and produce inflammatory factors such as interferon-α (IFN-α) and tumor necrosis factor (TNF) [20]. In turn, inflammatory factors can act as paracrine signals to activate STAT and NF-κB signaling pathways in carcinoma cells, thus supporting the malignant progression of gliomas [20,65]. In addition, GAP43 is an actin-associated protein primarily responsible for regulating the formation of neural axon growth cones [66,67]. In astrocytes, GAP43 appears to be associated with the formation and extension of cytomembrane protrusions. Osswald M et al. found that GAP43 expression is upregulated in astrocytes in the context of gliomas, extends cytomembrane protrusions with GFAP as a supportive backbone, connects with glioma cells via GAP43, and constitutes a functional and resistant malignant cellular network. This network allows for the buffering of lethal damage within individual cells, such as the high levels of calcium release accompanying endoplasmic reticulum/mitochondrial damage [68]. It improves the viability of glioma cells. Furthermore, Watson et al. discovered that GAP43 expression is upregulated in GFAP+ astrocytes, potentially enhancing GBM tumorigenicity by facilitating mitochondrial transfer from reactive astrocytes to tumor cells through direct contact [18]. Thus, the GJ-dependent glioma–astrocyte network could increase the tolerance of gliomas during high growth rates, significantly improving their survivability and malignancy. Interestingly, GAP43 can inhibit the activity of protein kinase and AKT signaling pathways in C6 glioma cells, demonstrating a tumor growth inhibitory effect. Typically, GAP43 expression is absent in glioma cells [69]. Thus, GAP43 can help distinguish glioma cells from reactive astrocytes.
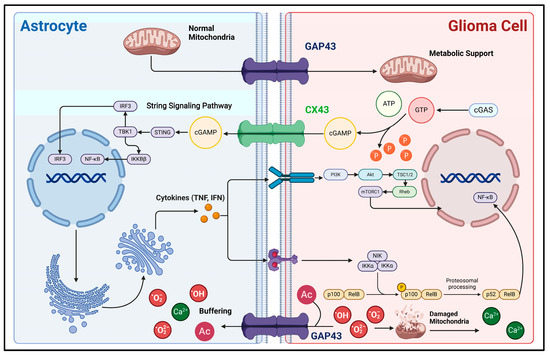
Figure 2.
Mechanisms of direct material and information interaction between glioma cells and astrocytes. This figure illustrates the material interactions and associated signaling pathways between astrocytes and glioma cells via direct cell-to-cell contact. CX43 and GAP43 are important gap junction proteins between glioma cells and astrocytes, acting as bridges for the exchange of substances and information. During the rapid proliferation of glioma cells, due to their high demand for energy, their metabolic mode shifts to aerobic glycolysis as the main process. Astrocytes can transport normal mitochondria into glioma cells through GAP43, supporting their energy metabolism [18]. Also, aerobic glycolysis can generate a large amount of reactive oxygen species (ROS). First, ROS can cause mitochondrial damage, which releases a large amount of calcium ions, leading to calcium overload in glioma cells and subsequent apoptosis [70]. Second, the excessive accumulation of ROS itself is lethal. Glioma cells can transport these lethal factors into the astrocyte buffer pool network through GAP43, thereby developing resistance to cell death [69]. In addition, direct cell-to-cell contact is involved in the process of astrocytes acquiring reactivity. The signaling molecule cGAMP is produced within glioma cells and then transported into astrocytes via CX43. This activates the downstream STING and NF-κB signaling pathways, enabling astrocytes to acquire reactivity [20]. In this figure, the arrow represents the direction of the transmission of substances and signaling molecules.
4.2. Molecular Transfer via Non-Direct Contact
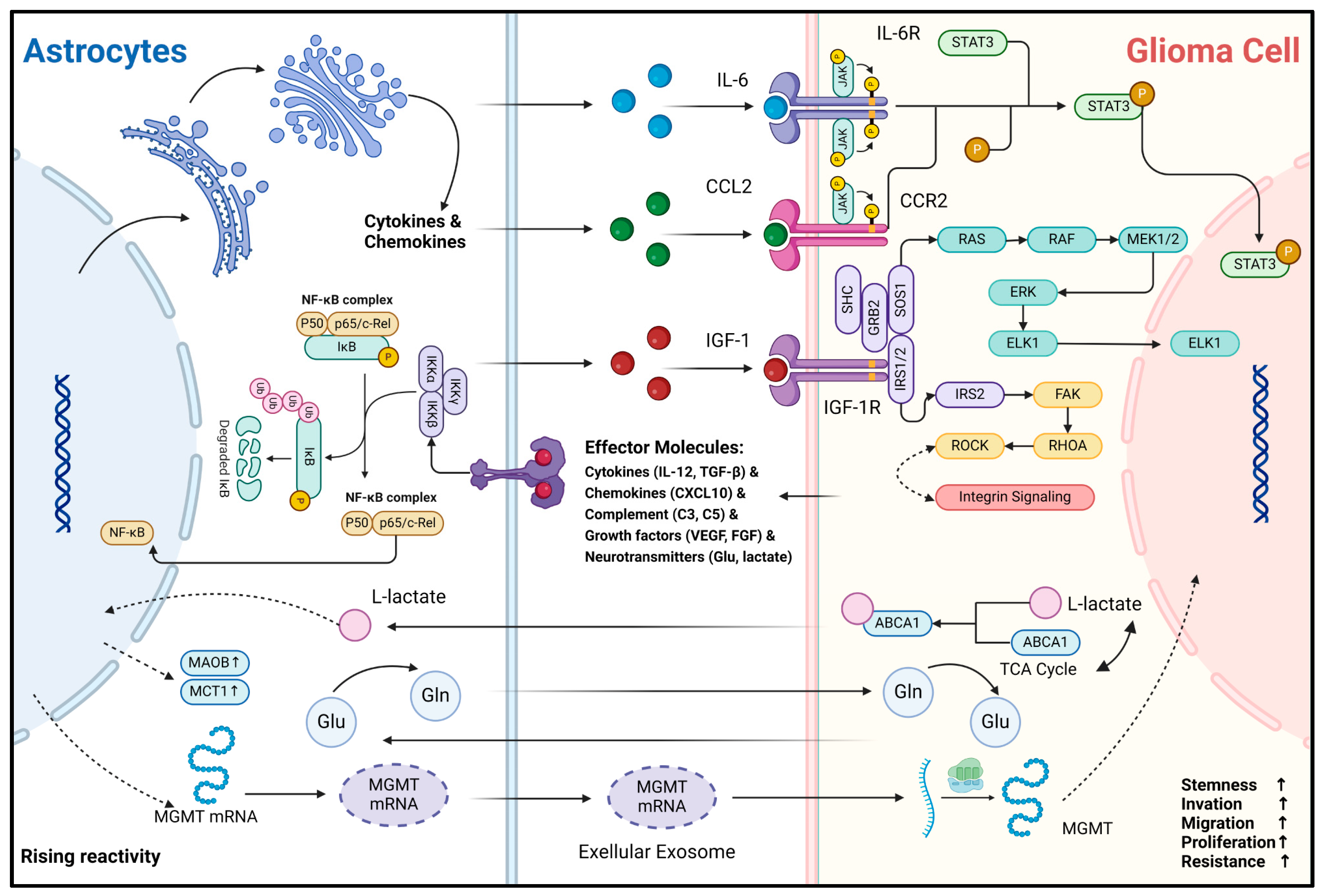
Another important biological function of astrocytes is their ability to secrete a diverse range of effector substances upon sensing abnormal CNS events. In CNS inflammatory diseases, reactive astrocytes have been extensively studied. Their effector molecules are broadly categorized as follows: cytokines (e.g., IL-1β, IL-6, TNF, IFN-γ, TGF-β) [10,12,71], chemokines (e.g., CCL2, CCL3, CCL5, CCL20, CXCL1, CXCL10) [72], complement cascade components (e.g., C3, C5, CFB) [8], growth factors (e.g., VEGFA, VEGFB, FGF, BMP1, NGF, BDNF, PDGFD) [73], and neurotransmitters (e.g., Glutamate, ATP) [74,75]. The available evidence suggests that effector molecule secretion profiles of reactive astrocytes overlap between gliomas and non-tumorigenic CNS inflammatory diseases [45,76]. These molecules can bind to receptors on the surface or inside glioma cells, which in turn modulate the transformation of their malignant phenotype (Figure 3). For example, Oushy et al. found that extracellular vesicle components in glioblastoma cell culture medium supernatants stimulated astrocyte reactivity, which in turn upregulated the release of molecules such as IL-12, IL-1A/B, CXCL10, and C5 [45]. These effector molecules can be directly recognized by glioma cells and play crucial roles in regulating their acquisition and maintenance of malignancy. IL-6 can bind to IL-6R on the surface of glioma cells and activate the downstream JAK2/STAT3 signaling pathway, enabling them to acquire the stemness and invasiveness phenotype [22]. IGF-1R is widely present and highly expressed in glioma cells [77]. Exogenous IGF-1 can enhance the invasive and migratory capacity of glioma cells by binding to IGF-1R and has been associated with resistance of gliomas to radiation, chemotherapy, and immunotherapy [25,77,78,79]. In addition, CCL20 can be released by astrocytes to assist gliomas in counteracting hypoxia by binding to CCR6 to activate the NF-κB pathway and to upregulate HIF-1 expression [24]. Astrocyte-derived CCL2 can bind CCR2 on glioma cells to maintain stemness characteristics by activating JAK2/STAT3-Notch signaling pathway [23]. The effector molecules represented by cytokines and chemokines are relatively stable and can be delivered to glioma cells by paracrine secretion or extracellular vesicle trafficking. Notably, RNA transport between cells is a recent hot research topic. These less stable molecules and intracellular components can also be delivered into glioma cells and functionalized via extracellular vesicles or exosomes. A recent study indicates that mRNA for O6-alkylguanine DNA alkyl transferase (MGMT) is significantly elevated in exosomes from reactive astrocytes, enabling glioma cells to resist temozolomide (TMZ)-induced apoptosis by sequestering these exosomes [80]. However, only the phenomenon that glioma-associated astrocytes can produce and secrete these specific effector molecules is mentioned in these studies. Considering the holistic concept of the glioma microenvironment, other microenvironmental components such as microglia may also be part of the extra source of these effector molecules like IL-6 and CCR2 [81,82]. Therefore, the primary source of these effector molecules must be critically considered and investigated in the future. Fortunately, the rise of single-cell and spatial transcriptome sequencing technologies has provided effective ways to explore this question.
Secondly, metabolic and nutritional support is one of the important physiological functions of astrocytes. Astrocytes can generate metabolites such as L-lactate and L-serine via glycolysis and shuttle them to neurons to maintain energy requirements [83]. Also, astrocytes recycle toxic metabolites and neurotransmitters such as fatty acids and glutamate produced by neurons to protect normal neuronal function during periods of enhanced activity [55,84]. In the context of gliomas, astrocytes still fulfill their role as “metabolic transit stations”. L-glutamine (Gln) is crucial for maintaining carbon and nitrogen balance in neural tissues, primarily supplied by astrocytes in the CNS [85]. Under Gln-deficient conditions, the growth rate of gliomas is significantly inhibited [86]. Tardito et al. found that in GBM cells, almost half of the glutamate produced via the glutamine–glutamate cycle is secreted, but it does not enter the tricarboxylic acid (TCA) cycle. Instead of producing Gln for consumption, astrocytes synthesize Gln by uptake of glutamate and subsequently become the major provider of Gln for GBM growth metabolism [46]. Similarly, Perelroizen et al. found that reactive astrocytes can metabolically support GBM by delivering cholesterol extracellularly via ABCA1 [27]. Conversely, acetic acid, an intermediate product of GBM metabolism, upregulates the expression of MAO-B and MCT1 upon astrocyte uptake, in turn promoting the reactivity and proliferation of neighboring astrocytes [21]. In addition, glioma cells retain part of the metabolic characteristics of astrocytes, such as functioning in an aerobic glycolytic manner and producing the intermediate metabolite lactate. Under hypoxia and glucose deficiency, lactate can modulate astrocytes and reduce their inflammatory response [87]. Although the study was conducted in ischemic stroke models, the results align with our understanding of the glioma microenvironment, characterized by oxygen and nutrient deficiencies and immunosuppression. This gives us a hint that, in addition to the complementarity of metabolites, there is a synergy between reactive astrocytes and glioma cells in the process of microenvironment metabolic remodeling. The driving forces behind these metabolic remodeling and interaction processes in the oncogenesis and progression of gliomas, as well as the specific mechanisms by which subsequent effects are generated, deserve further in-depth exploration.
In summary, reactive astrocytes and glioma cells engage in various material and information exchange pathways, facilitating a mutual support that plays a crucial role in the mechanism of the malignant progression of gliomas.
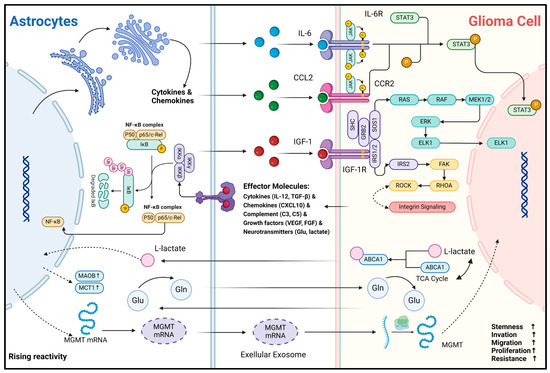
Figure 3.
Mechanisms of non-direct material and information interaction between glioma cells and astrocytes. This figure illustrates the interaction and signaling mechanisms between astrocytes and glioma cells via non-direct contact. As the reactivity of astrocytes increases, the expression of molecules such as MAOB and MCT1 rises, accompanied by metabolic changes in substances like L-lactate, glutamic acid (Glu), and glutamine (Gln). Gln can be synthesized by astrocytes and transported into glioma cells to support their metabolism. The metabolite of Gln, Glu, can then be transported back into astrocytes for the resynthesis of Gln, thus completing the Glu-Gln metabolic cycle [46]. In astrocytes, cytokines and chemokines are produced, and the NF-κB complex plays a crucial regulatory role [88]. Cytokines and chemokines secreted by astrocytes, such as IL-6, CCL2, and IGF-1, can act on the corresponding receptors (IL-6R, CCR2, IGF-1R, etc.) on glioma cells, activating downstream signaling pathways like JAK-STAT3 and RAS-RAF-MEK1/2-ERK and involving integrin signaling [22,23,45]. Glioma cells regulate their biological behaviors, including stemness, invasion, migration, proliferation, and resistance, through these signaling pathways. In addition, extracellular exosomes are involved in the information transfer between the two; for example, MGMT mRNA can be transported from reactive astrocytes to glioma cells via extracellular vesicles, contributing to the development of resistance to TMZ chemotherapy in glioma cells [80]. In this figure, the arrow represents the direction of the transmission of substances and signaling molecules.
Figure 3.
Mechanisms of non-direct material and information interaction between glioma cells and astrocytes. This figure illustrates the interaction and signaling mechanisms between astrocytes and glioma cells via non-direct contact. As the reactivity of astrocytes increases, the expression of molecules such as MAOB and MCT1 rises, accompanied by metabolic changes in substances like L-lactate, glutamic acid (Glu), and glutamine (Gln). Gln can be synthesized by astrocytes and transported into glioma cells to support their metabolism. The metabolite of Gln, Glu, can then be transported back into astrocytes for the resynthesis of Gln, thus completing the Glu-Gln metabolic cycle [46]. In astrocytes, cytokines and chemokines are produced, and the NF-κB complex plays a crucial regulatory role [88]. Cytokines and chemokines secreted by astrocytes, such as IL-6, CCL2, and IGF-1, can act on the corresponding receptors (IL-6R, CCR2, IGF-1R, etc.) on glioma cells, activating downstream signaling pathways like JAK-STAT3 and RAS-RAF-MEK1/2-ERK and involving integrin signaling [22,23,45]. Glioma cells regulate their biological behaviors, including stemness, invasion, migration, proliferation, and resistance, through these signaling pathways. In addition, extracellular exosomes are involved in the information transfer between the two; for example, MGMT mRNA can be transported from reactive astrocytes to glioma cells via extracellular vesicles, contributing to the development of resistance to TMZ chemotherapy in glioma cells [80]. In this figure, the arrow represents the direction of the transmission of substances and signaling molecules.
5. Reactive Astrocytes and Remodeling of the Glioma Microenvironment
Astrocytes have multiple functions in the normal state, such as regulating water homeostasis, participating in the construction of the blood–brain barrier, constructing the extracellular matrix, and regulating angiogenesis and hemodynamics [89]. In the context of glioma, the ability of astrocytes to maintain homeostasis is disrupted, which may be related to the formation of the peritumoral edema zone, the development of tumor blood vessels, the remodeling of the extracellular matrix associated with invasion, and the breakdown of the blood–brain barrier. Secondly, astrocytes can secrete a variety of cytokines, which may be involved in the recruitment of cells that make up the microenvironment (Figure 4). Therefore, the potential mechanisms of the interaction between astrocytes and the glioma microenvironment deserve further attention.
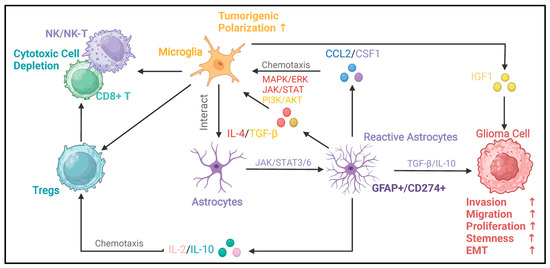
Figure 4.
Oncogenic immunoregulatory functions of reactive astrocytes in the glioma microenvironment. This figure illustrates the oncogenic immunoregulatory functions of glioma-reactive astrocytes. Reactive astrocytes can secrete the chemokine CCL2 and CSF1 [27]. By activating the MAPK/ERK, JAK/STAT, and PI3K/AKT signaling pathways, they drive the polarization of microglia toward pro-tumor phenotypes [44]. Microglia with a pro-tumor phenotype can further promote the malignant progression of glioma cells through factors such as IGF1. Conversely, microglia can secrete factors such as TGF-β and IL-10 [13,14,44]. By activating signaling pathways like JAK/STAT3/6, these factors enable astrocytes to acquire reactivity. Reactive astrocytes can secrete TGF-β and IL-10, which promote the invasion, migration, proliferation, stemness, and epithelial–mesenchymal transition (EMT) of glioma cells [90,91]. In addition, cytokines such as IL-2 and IL-10 secreted by reactive astrocytes can chemotaxis Tregs [24,28,44,45]. Tregs and microglia can interact with cytotoxic cells (NK/NK-T cells, CD8+ T cells), leading to the depletion of cytotoxic cells and immune-suppressive microenvironment [92,93]. In this figure, the arrows between different types of cells indicate the interaction relationships among the cells. The upstream of the arrow represents the regulator, and the downstream represents the responder. The arrow between astrocytes and reactive astrocytes represents the process and direction of polarization. The signaling molecules and signaling pathways in the middle describe the key signaling molecules and signaling pathways involved in these regulatory processes.
5.1. Reactive Astrocytes and Glioma Angiogenesis
During normal CNS development, astrocytes play an important role in the formation and development of blood vessels. Astrocytes can provide a growth framework for vascular endothelial cells and serve as a template for new blood vessels [94,95]. The disruption of the astrocyte template can lead to the formation of abnormal blood vessels. For example, selective interference with astrocytes in the retina can cause abnormal blood vessel networks [96]. To support their function as the angiogenesis scaffold, multiple cell adhesion molecules are present on the surface of astrocytes, such as cadherins, integrins, and laminins [97]. These adhesion molecules may provide potential sites for tumor angiogenesis. Notably, glioma cells retain characteristics like those of astrocytes and can express similar adhesion molecules on the surface. However, unlike astrocytes, tumor cells can rely on surface adhesion molecules to establish blood supply pathways by the unique mechanism of vasculogenic mimicry (VM). For example, VE-Cadherin is regulated by the transcription factor FOXK1 and is highly expressed on the surface of glioma cells. It plays a crucial role in VM formation [98]. β8 integrin (ITGB8) can promote the formation of VM through the Smad2/3-RhoA signaling pathway in GBM [99]. Since VM provides channels for cancer invasion and metastasis, these surface adhesion molecules may be crucial pathways for the progression and acquisition of blood supply support of gliomas [100]. Secondly, astrocytes can secrete pro-angiogenic factors, including VEGF, FGF, and angiopoietin [101,102,103,104]. Under pathological conditions, the expression patterns of these factors will change. For example, under hypoxic conditions, HIFs can stimulate astrocytes to upregulate VEGF expression [105]. In ischemic stroke, HIF-1α-related signals can prompt reactive astrocytes to polarize towards the A2 phenotype. This can increase the expression of VEGF for promoting the formation of new blood vessels and protect the brain tissue from ischemia and hypoxia damage [106]. In the context of glioma, similar changes have also been reported. Vasiliki et al. found that reactive astrocytes in GBM tissues are localized near the peri-necrotic regions of HIF-2α-expressing cells and exhibit a robust hypoxic response. This is driven by HIF-2α, manifested as an elevation in the expression of hypoxia-related cytokines, including TGF-β1, IL-3, angiopoietin, VEGFA, and IL-1α [107]. In addition, some pro-angiogenic factors are also involved in the process of VM formation. For example, VEGF is also an important regulatory molecule of VM, and its expression is regulated by HIFs in the hypoxic microenvironment [108]. In summary, various evidence suggests that reactive astrocytes may directly participate in the regulation of glioma angiogenesis (Figure 5A). However, there are no direct experimental studies that have achieved a reduction in blood supply and inhibition of progression by intervening with glioma-reactive astrocytes. Angiogenic factors like VEGF are likewise expressed in glioma cells, microglia, etc. Whether the construction and remodeling of the glioma blood supply is dominated by astrocytes remains debatable. These issues deserve further exploration in the future.
5.2. Reactive Astrocytes and Disturbance of Water Homeostasis in Glioma
Astrocytes in the brain parenchyma are the main cell types expressing aquaporins. Aquaporin 4 (AQP4) is one of the important aquaporins on astrocytes, which is mainly enriched in the end-feet, located at the blood–spinal cord barrier and the blood–brain barrier, regulating water exchange on both sides [109,110]. It plays an important role in maintaining the stability of the blood–brain barrier, cell volume, and extracellular space volume of astrocytes [110]. Under pathological conditions, the expression and distribution of AQP4 in reactive astrocytes will be altered. The glymphatic system promotes the reflux of cerebrospinal fluid and waste clearance in the brain during sleep through perivascular channels supported by glial cells [111,112]. In patients with Parkinson’s disease (PD), the localization of AQP4 in the end-feet of astrocytes is reduced (AQP4 depolarization), manifested as metabolic disorders in cerebrospinal fluid reflux and neurotransmitter clearance related to the impaired glymphatic system [113]. This exacerbates the loss of dopaminergic neurons. Similarly, AQP4 depolarization also occurs in patients with Alzheimer’s disease (AD) and normal-pressure hydrocephalus. It is characterized by the accumulation of cerebral metabolic waste and amyloid-β plaques, as well as an increase in perivascular reactive astrocytes, which further accelerates the progression of cognitive impairment [114]. In a stroke model with ischemia and hypoxia, AQP4 was overexpressed in reactive astrocytes around the infarct area. Targeted inhibition of AQP4 could significantly alleviate post-infarct cerebral edema [115]. Cognitive impairment, as well as increased intracranial pressure and headaches caused by peritumoral tissue edema, are also common clinical symptoms in glioma. Therefore, an abnormal expression and localization of AQP4 may exist in glioma and form part of the mechanism underlying the generation of clinical symptoms (Figure 5B). Existing evidence only mentions the increased expression of AQP4 in glioma-reactive astrocytes but does not further clarify its specific mechanisms in regulating the flow of edema fluid in gliomas [116]. These issues deserve more profound and extensive research in the future.
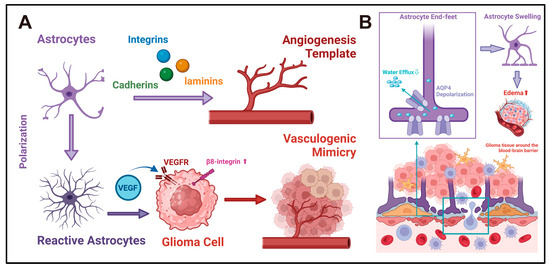
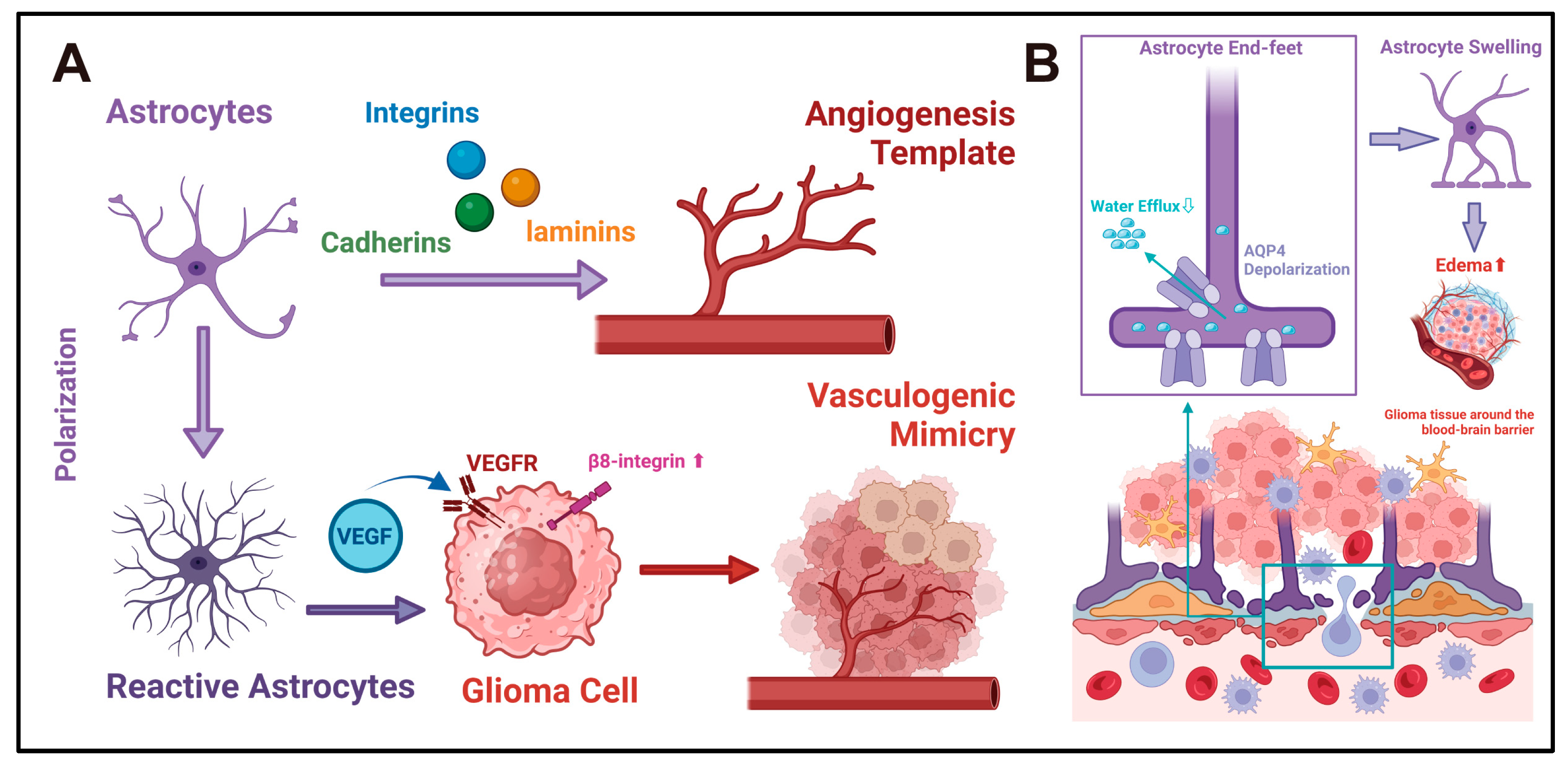
Figure 5.
(A) Reactive astrocytes and angiogenesis in gliomas: Astrocytes may influence glioma angiogenesis via two potential mechanisms. First, astrocytes express diverse adhesion molecules, such as cadherins, integrins, and laminins, which offer attachment sites for endothelial cells, thereby serving as the template and framework for CNS angiogenesis [90,91]. Second, reactive astrocytes secrete angiogenic factors like VEGF. By facilitating the VM process, they may supply additional blood sources and invasion pathways for gliomas [101,102,103,104,107]. (B) Astrocytes and water homeostasis in gliomas: AQP4 is the primary aquaporin in astrocytes and is localized to the astrocytic end-feet. It plays a crucial role in regulating cerebrospinal fluid circulation at the blood–brain barrier interface, thereby maintaining the water homeostasis of CNS [109,110]. In the pathological context, the depolarization of AQP4 occurs at the end-feet of astrocytes [113]. This impedes the efflux of cerebrospinal fluid across the blood–brain barrier. As a result, the volume of astrocytes increases, and cerebrospinal fluid accumulates in the interstitial space. This might be one of the mechanisms underlying the development of glioma-associated edema. In this chart, the large arrows represent the direction of the functional evolution of cells or the interactions between cells. The signaling molecules in the middle of the arrows represent the key regulatory factors in this process. The small arrows represent the sites of action of the signaling molecules or the direction of the flow of substances.
Figure 5.
(A) Reactive astrocytes and angiogenesis in gliomas: Astrocytes may influence glioma angiogenesis via two potential mechanisms. First, astrocytes express diverse adhesion molecules, such as cadherins, integrins, and laminins, which offer attachment sites for endothelial cells, thereby serving as the template and framework for CNS angiogenesis [90,91]. Second, reactive astrocytes secrete angiogenic factors like VEGF. By facilitating the VM process, they may supply additional blood sources and invasion pathways for gliomas [101,102,103,104,107]. (B) Astrocytes and water homeostasis in gliomas: AQP4 is the primary aquaporin in astrocytes and is localized to the astrocytic end-feet. It plays a crucial role in regulating cerebrospinal fluid circulation at the blood–brain barrier interface, thereby maintaining the water homeostasis of CNS [109,110]. In the pathological context, the depolarization of AQP4 occurs at the end-feet of astrocytes [113]. This impedes the efflux of cerebrospinal fluid across the blood–brain barrier. As a result, the volume of astrocytes increases, and cerebrospinal fluid accumulates in the interstitial space. This might be one of the mechanisms underlying the development of glioma-associated edema. In this chart, the large arrows represent the direction of the functional evolution of cells or the interactions between cells. The signaling molecules in the middle of the arrows represent the key regulatory factors in this process. The small arrows represent the sites of action of the signaling molecules or the direction of the flow of substances.
5.3. Reactive Astrocyte–Microglia Interactions
Unlike other tissues, the CNS has unique immunity exemption mechanisms due to the presence of the blood–brain barrier. This results in the macrophage/microglia cells residing in the CNS being the predominant immune cell component in the general state. Thus, microglia may be among the first responders to abnormal events in the CNS [117,118]. Secondly, astrocytes are also prevalent in the CNS, so their immunomodulatory function has attracted attention. However, during the construction of reactive astrocyte models in the context of inflammation, it was found that although astrocytes express TLR4, the lack of intracellular response mechanisms (e.g., MYD88) prevented LPS stimulation alone from directly rendering astrocytes reactive [119,120]. However, cytokine-rich supernatants of microglia receiving LPS stimulation can induce A1 astrocytes in murine model [8]. This landmark discovery has directed attention to microglia–astrocyte interaction mechanisms. Recent studies indicate that astrocytes are crucial support cells for the growth and survival of microglia. For example, astrocyte-derived IL-34, TGF-β, and cholesterol molecules support branching microglia survival in vitro and maintain microglia homeostasis in the CNS [121]. In the context of CNS inflammation, microglia are important for astrocyte activation. In a sepsis model, microglia can polarize reactive astrocytes by releasing IL-1α, TNF-α, and C1q, which activates caspase-2/-3 signaling in neurons, leading to apoptosis and neurological damage [8]. In contrast, microglia can also signal astrocytes through molecules like TGF-β, which helps balance and protect against reperfusion injury in ischemic stroke [122].
Compared to non-tumor inflammatory diseases, the acquisition of astrocyte reactivity in the context of glioma possesses its unique mechanism. Glioma cell secretory products can directly induce GFAP-positive reactive astrocytes, and models of glioma-associated reactive astrocytes have been constructed in multiple studies based on this understanding [20,45,80]. Microglia primarily facilitate the responsiveness of astrocytes. Henrik et al. revealed that the formation of GFAP+CD274+-reactive astrocytes, which create an immunosuppressive microenvironment, relies on activating the JAK/STAT signaling pathway. The depletion of microglia in a glioma model in human brain slices resulted in a significant decrease in the activity of the astrocyte JAK/STAT signaling pathway, which was accompanied by a downregulation of GFAP and CD274 expression [28]. Also, reactive astrocytes can influence the microglia phenotype. Perelroizen R et al. pointed out that reactive astrocytes can recruit microglia/TAMs and give them a pro-tumorigenic phenotype by upregulating the expression of CCL2 and CSF1 [27]. In addition, a multilateral cytokine interaction network exists among microglia, astrocytes, and tumor cells. Yao et al. found that medulloblastoma stimulated IL-4 secretion from reactive astrocytes in a STAT6-dependent manner. Microglia receiving IL-4 signals upregulate the activity of the JAK/STAT and MAPK/ERK signaling pathways and secrete IGF1, promoting tumor cell growth [44]. In summary, microglia could be important regulators of the acquisition of responsiveness by astrocytes and functionally synergize or complement with astrocytes in promoting the malignant progression of gliomas. These findings provide new insights and ideas for the research of the glioma microenvironment.
5.4. Reactive Astrocyte–Circulating Immune Cell Interactions
During the formation of gliomas, many circulating immune cells are recruited into the tumor tissue with the disruption of the blood–brain barrier. Cytotoxic cells, represented by CD8+ T cells and NK cells, exert a major tumor-lysogenic capacity. However, gliomas evolve to initiate mechanisms of immune escape, frequently manifest as cytotoxic cell depletion. This largely depends on the widespread presence of immunosuppressive effector molecules in the glioma microenvironment [123]. Among the available evidence, glioma-reactive astrocytes can secrete a variety of cytokines and chemokines, including IGF1, CCL20, TGF-β, IL-10, IL-12, IL1A/B, IL-2, CXCL10, and G-CSF [24,28,44,45]. These cytokines and chemokines proved to be engaged in recruiting and polarizing specific circulating immune cells in glioma. For example, IL-10 recruits Treg cells and enhances their function, promoting depletion of T cells within the tumor [92,93]. Similarly, IL-2 has been shown to activate Treg cells residing in the CNS and function to reverse neuroinflammation. TGF-β is an important immunosuppressive molecule in the glioma microenvironment. TGF-β can induce the polarization of TAMs towards an M2-like phenotype by activating the PI3K/AKT signaling pathway, and it can also facilitate glioma cells’ evasion from NK cells through the αv integrin-mediated TGF-β signaling pathway [13,14]. Thus, based on circumstantial evidence, information interaction channels between reactive astrocytes and circulating immune cells seem to exist. However, there is no direct evidence to support this hypothesis in the context of glioma. These mechanisms remain need to be elucidated and refined in the future.
5.5. Reactive Astrocyte–Extracellular Matrix Interactions
CNS injury, inflammation, or tumor lesions are often accompanied by a remodeling process of the extracellular matrix (ECM). Astrocytes serve an important secretory function in the CNS and are a major source of ECM structure molecules [124]. In the pathological state, the acquisition of reactivity by astrocytes is followed by a marked change in the secretory function of ECM. After CNS injury, reactive astrocytes may form the glial scar by changing ECM secretion patterns, potentially serving as a protective precaution to limit further injury and inflammation spread [125,126]. Similarly, peri-cancerous astrocytes can form astrogliosis proliferation bands and encase glioma tissue. This may be related to specific pathologic changes in gliomas. For example, glioma stem cells can upregulate the expression of MAO-B and MCT1 in astrocytes via the metabolite acetate or inhibit the activity of the p53 signaling pathway in astrocytes to induce astrogliosis [21,127]. In addition, impaired blood–brain barrier function causes the deposition of plasma-derived fibrinogen and fibronectin. These exogenous ECM proteins can activate the TGF-β signaling pathway in astrocytes, which in turn alters the expression and secretion of ECMs such as chondroitin sulfate [90,91]. However, whether the astrogliosis bands can limit glioma invasion and metastasis is currently unsettled. Among the current findings, the expression and secretion patterns of reactive astrocytes seem to indicate that they can contribute to glioma progression through ECM reorganization. VIM is one of the important upregulated markers of reactive astrocytes [128]. Also, VIM is an important marker for mesenchymal subtypes of glioma tissues, which are closely related to EMT, invasion, and metastatic processes [129,130]. Lu et al. found that tumor cells could induce astrocytes to acquire reactivity through the Wnt/β-catenin signaling pathway, as evidenced by a decrease in E-calmodulin expression and an increase in the expression of VIM and MMPs. They hypothesized that the conversion of astrocytes to a mesenchymal subtype could assist tumor cells in catabolizing the extracellular matrix, which in turn promotes invasion and metastasis [131]. Le et al. found that reactive astrocytes provide precursor proteins to MMP2, which are cleaved via the activation of fibrinolytic enzymes by the tumor cells to catabolize the extracellular matrix in the form of active MMP2 [17]. Thus, the astrogliosis bands in gliomas seem not to be dense but rather full of “channels”. Reactive astrocytes that acquire a mesenchymal-like phenotype may be one of the facilitators of migration and invasion.
Overall, reactive astrocytes are important regulators of the microenvironment and can potentially promote the malignant progression of gliomas.
6. Reactive Astrocytes in Glioma Treatment
Currently, the standard treatment for gliomas is surgical resection supplemented by TMZ chemotherapy and radiotherapy [132]. However, the repeated tendency of gliomas to recur and their susceptibility to develop resistance to radiation/chemotherapy pose a great challenge for their clinical management. Although novel glioma treatment strategies such as anti-angiogenic therapies, immunotherapies, epigenetic therapies, oncolytic viral therapies, and gene therapies have shown promising performance in preclinical trials, their actual performance in clinical trials is poor, and they are accompanied by risk or uncertainty of efficacy [133]. Here, we address the potential mechanisms and feasible coping strategies for reactive astrocytes to assist gliomas in generating radiation/chemotherapy resistance. At the same time, we discuss the feasibility of astrocyte-targeted therapies in gliomas and future directions based on advances in astrocyte-targeted therapies in CNS inflammatory diseases.
6.1. Reactive Astrocytes and Glioma Radiotherapy/Chemotherapy Resistance
As we mentioned above, intensive material and information exchange exists between reactive astrocytes and glioma cells, leading to the development of resistance to radiotherapy and chemotherapy through various mechanisms. Astrocytes can connect with gap junction connections on the glioma surface through CX43, significantly reducing apoptosis in glioma cells induced by the temozolomide and vincristine [29]. The mRNA of MGMT can be delivered from reactive astrocytes to tumor cells via exosomes, which enhances their resistance to TMZ-induced apoptosis [80]. Also, upregulated expression of IL1-β by reactive astrocytes can stimulate the transformation of glioma-initiating cells to mesenchymal-like subtypes, leading to their therapeutic resistance [134]. In turn, radiation/chemotherapy can induce abnormally reactive astrocytes. Fletcher-Sananikone E et al. pointed out that stromal cells in the glioma microenvironment can acquire a senescence-associated secretory phenotype upon exposure to ionizing radiation, and this is particularly evident in astrocytes. Under such conditions, activated reactive astrocytes upregulate the expression of senescence factors such as CDKN1A, HGF, and RTK, increasing the invasiveness of glioma cells by activating the MET signaling pathway [32]. Berg et al. found that ionizing radiation can induce reactive astrocytes, which can promote glioma stem cell survival by upregulating transglutaminase 2 (TGM2) [30]. Targeting TGM2 can significantly reduce tumor recurrence and post-radiotherapy cancer volume [30]. Also, the TGM2-related Gln-Glu cycle facilitates metabolite exchange between gliomas and astrocytes [46]. Moreover, the transfer of mitochondria from stromal cells to glioma cells might be related to the emergence of chemoresistance. This phenomenon has been reported in a study of microglia and GBM [135]. Similarly, mitochondrial transfer also occurs between reactive astrocytes and glioma cells [18]. Interestingly, Sun C et al. implanted astrocyte-derived mitochondria into gliomas, which instead increased their sensitivity to radiotherapy [135,136]. Thus, this topic has become controversial. We believe that a transient increase in the number of mitochondria within glioma cells may be beneficial for the radiotherapy effect. This is because mitochondria damaged by ionizing radiation can release apoptotic signals, which are conducive to the clearance of cancer cells. However, continuous mitochondrial delivery may be harmful, as it may endow the remaining glioma cells with stronger proliferative and invasive capabilities. In summary, these results have prompted us to wonder whether other mechanisms are involved in the exchange of substances and information between reactive astrocytes and gliomas that contribute to the development of therapeutic resistance. Can targeting these pathways enhance the effectiveness of radiation and chemotherapy, reduce glioma recurrence, and extend patient survival? Future research needs to address these questions.
6.2. Potential Strategies for Targeting Reactive Astrocytes in Gliomas
The key to effectively targeting reactive astrocytes is to precisely manipulate their phenotype in the microenvironment and peritumor, freeing them from their “pro-tumorigenic” state or enabling them to inhibit glioma growth. Currently, abundant information exists on reversing the astrocyte phenotype in CNS inflammatory and degenerative diseases, which can provide new insights into targeting therapeutic strategies for gliomas. Firstly, the NF-κB signaling pathway is crucial for regulating astrocyte reactivity [137]. Strategies targeting this pathway can significantly reduce the generation of GFAP+ reactive astrocytes and alleviate inflammations in abnormal CNS events like stroke, cerebral hemorrhage, Alzheimer’s disease, and spinal cord injury [88,138,139,140]. Secondly, intervention in reactive astrocytes can lead to a reversal of their pro/anti-inflammatory phenotype. For example, nicotinamide adenine dinucleotide-dependent deacetylase sirtuin-1 (SIRT1) is a crucial regulator of the pro-inflammatory response in reactive astrocytes. Knockdown of SIRT1 in astrocytes can reduce T-cell infiltration in the CNS and elevate the ratio of macrophages/microglia with IL-10 secretion, manifested in the conversion of reactive astrocytes from pro-inflammatory to anti-inflammatory phenotype [141]. Although these studies aimed to suppress the aberrant inflammatory response in CNS, they offer inspiration for the development of targeted therapies for glioma-reactive astrocytes. Under the selection of appropriate molecules or pathways, artificial intervention into reactive astrocytes is achievable. This might potentially fulfill the conversion of glioma-reactive astrocytes possessing tumor-promoting capabilities into those astrocytes capable of normally reacting to anti-tumor immunity. To fulfill this vision, we need to understand the generation and inside regulatory mechanisms of glioma-reactive astrocytes. Research on this topic will be valuable for the development of astrocyte-targeted therapies.
Second, regarding effects, extensive material information interaction pathways exist between astrocytes and glioma cells. Key enzymes of the lactate, acetate, and Gln/Glu metabolic cycles and gap junction proteins such as CX43 and GAP43 that act as bridges of material and information can serve as potential targets for intervention within astrocytes [18,19,20,21,46,66,69,83]. Among them, the exploration of blockade therapies targeting the metabolic loops between gliomas and microenvironment cells has received much attention. The glutamine–glutamate cycle is a suitable site. Intervening in its synthesis and transport processes may be effective for gliomas. For example, Zhong et al. mentioned that alanine-serine-cysteine transporter 2 (ASCT2) is a key glutamine transporter. Inhibiting the activity of ASCT2 can significantly reduce the uptake of glutamine by glioma cells, directly inhibit the growth of GBM, and decrease its resistance to TMZ therapy [142]. TGM2 is a key enzyme in glutamate metabolism. Inhibiting the activity of TGM2 can significantly enhance the sensitivity of GBM to radiotherapy [30,143]. The enzyme glutamine synthetase (GS), which is abundant in astrocytes, catalyzes the conversion of glutamate to glutamine. Also, glutamine is related to the onset of glioma-related epilepsy [144]. Targeting the glutamate–glutamine cycle holds the potential not only to effectively impede the growth of gliomas but also to substantially reduce the incidence of epilepsy. This is beneficial for enhancing the quality of life for patients. Similarly, targeted therapies against the AQP4 molecule may also have the effect of inhibiting the growth of gliomas and improving clinical symptoms. As mentioned above, the dysregulation of the AQP4 molecule may be related to cerebral edema, increased intracranial pressure, headache, and cognitive impairment. In gliomas, AQP4 has additional pathophysiological functions beyond its role as water channels. These functions are associated with glioma invasion, angiogenesis, and malignant progression [145,146]. Therefore, AQP4 is also a promising target for clinical intervention. In addition, targeted therapy against the angiogenic capacity of glioma-reactive astrocytes is also a good option. VM can facilitate gliomas in obtaining blood supply and initiating invasion [98,99]. Despite cell adhesion molecules are important in the process of VM, VEGF signaling is indispensable in the formation of blood flow channel-like structures by cancer cells [100,108]. Reactive astrocytes are an important source of VEGF [107]. Inhibiting their release of VEGF may be effective in the treatment of gliomas. Table 1 compiles and presents the potential glioma treatment strategies currently reported, which center around the intervention of astrocytes. However, these studies may still be in the stage of preclinical research or ongoing clinical trials, and their specific mechanisms, as well as their actual performance and safety, remain to be further explored and evaluated. It is also unclear whether altering astrocyte metabolism impacts normal neuronal functions or whether shifting anti-inflammatory phenotypes to pro-inflammatory ones could hasten the recruitment of pro-tumorigenic immune cells to the microenvironment. These issues need to be identified and overcome in the future.

Table 1.
Currently reported glioma treatment strategies involving astrocyte targeting.
Overall, reactive astrocytes have non-negligible biological significance in microenvironment formation, metabolic regulation, pro-carcinogenic signaling, and acquisition of radiotherapy/chemotherapy resistance in gliomas. Targeting reactive astrocytes is a novel and promising strategy for the clinical management of gliomas that warrants further exploration in the future.
7. Conclusions
Astrocytes, an important component of the glioma microenvironment, are increasingly recognized as significant drivers of glioma progression. Reactive astrocytes can directly interact with tumor cells, enhancing malignancy and therapeutic resistance, and can also promote immune evasion, invasion, and metastasis through microenvironment remodeling. However, the pathophysiological mechanisms associated with reactive astrocytes are relatively advanced in non-tumor diseases, but there are still many imperfections in the context of glioma. Also, there are still large blanks regarding the heterogeneity of reactive astrocytes in the glioma microenvironment. Further understanding of the interacting mechanisms between astrocytes and gliomas will open new avenues for translational medicine and more meaningful clinical treatments for gliomas.
Author Contributions
J.W. (Jiasheng Wu), R.L., J.W. (Junwen Wang) and Y.M. collected and organized the documentation in this work, and J.W. (Jiasheng Wu) authored the manuscript. H.Z. and R.L. plotted the graphs in this work, and C.Y. and K.S. revised the manuscript and graphs. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Key R&D Program of China, MOST (2023YFC2510000), and the National Key R&D Program of China (2022YFC2403905); the National Natural Science Foundation of China (82403476); the Hubei Natural Science Foundation (2023AFB135); the China Postdoctoral Science Foundation (2022M711253); and the Wuhan Science and Technology Major Project (2021022002023426).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| GBM | glioblastoma |
| LGG | lower-grade glioma |
| GFAP | glial fibrillary acidic protein |
| TGF-β | transforming growth factor-β |
| SPARC | acid secretory protein |
| snRNA-seq | single nuclear RNA sequencing |
| scRNA-seq | single-cell RNA sequencing |
| CNS | central nervous system |
| GJs | gap junctions |
| IFN-α | interferon-α |
| TNF | tumor necrosis factor |
| MGMT | O6-alkylguanine DNA alkyl transferase |
| TMZ | temozolomide |
| Gln | L-glutamine |
| Glu | glutamate |
| TCA | tricarboxylic acid |
| EMT | epithelial–mesenchymal transition |
| GSCs | glioma stem cells |
| VM | vasculogenic mimicry |
| ITGB8 | β8 integrin |
| GS | glutamine synthetase |
| AQP4 | aquaporin 4 |
| ROS | reactive oxygen species |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| ECM | extracellular matrix |
| TGM2 | transglutaminase 2 |
| OPC-like | oligodendrocyte progenitor cell-like |
| NPC-like | neuronal progenitor cell-like |
| AC-like | astrocyte-like |
| MES-like | mesenchymal-like |
| VIM | vimentin |
| ASCL1 | achaete-scute complex-like 1 |
| EGFR | epidermal growth factor receptor |
| OLIG2 | oligodendrocyte transcription factor 2 |
| MKI67 | proliferating cell nuclear antigen Ki-67 |
| PTEN | phosphatase and tensin homolog |
| TP53 | tumor protein p53 |
| RB1 | retinoblastoma 1 |
| SOX2 | sex-determining region Y-box 2 |
| S100B | s100 calcium-binding protein B |
| S100A10 | s100 calcium-binding protein A10 |
| C1q | complement component 1 q subcomponent |
| NES | neuroepithelial stem cell protein |
| VIM | vimentin |
| C3 | complement component 3 |
| MMP-2 | matrix metallopeptidase 2 |
| PI3K | phosphatidylinositol 3 -kinase |
| Akt | protein kinase B |
| Src kinase | Rous sarcoma virus-related tyrosine-protein kinase src |
| cGAMP | cyclic guanosine monophosphate-adenosine monophosphate |
| STRT1 | nicotinamide adenine dinucleotide-dependent deacetylase sirtuin-1 |
| ASCT2 | alanine-serine-cysteine transporter 2 |
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hara, A.; Okayasu, I. Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: Correlation with angiogenesis and prognostic significance. Acta Neuropathol. 2004, 108, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, J.; Mittelbronn, M.; Nguyen, T.D.; Meyermann, R.; Beschorner, R. WT1 expression distinguishes astrocytic tumor cells from normal and reactive astrocytes. Brain Pathol. 2008, 18, 344–353. [Google Scholar] [CrossRef]
- Tanabe, K.; Matsushima-Nishiwaki, R.; Yamaguchi, S.; Iida, H.; Dohi, S.; Kozawa, O. Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J. Neuroinflamm. 2010, 7, 16. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón, Y.C.S.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Q.; Li, L.; Chen, K.; Yang, J.; Dixit, D.; Gimple, R.C.; Ci, S.; Lu, C.; Hu, L.; et al. β2-Microglobulin Maintains Glioblastoma Stem Cells and Induces M2-like Polarization of Tumor-Associated Macrophages. Cancer Res. 2022, 82, 3321–3334. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Zhu, H.; Liu, D.; Chen, Z.; Zhang, X.; Guo, Z.; Dong, M.; Wan, L.; Zhang, P.; Liu, G.; et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin αvβ5-Src-Stat3 signaling. Theranostics 2022, 12, 4221–4236. [Google Scholar] [CrossRef]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of astrocytes by glioma cells to facilitate invasiveness: A mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J. Neurosci. 2003, 23, 4034–4043. [Google Scholar] [CrossRef]
- Watson, D.C.; Bayik, D.; Storevik, S.; Moreino, S.S.; Sprowls, S.A.; Han, J.; Augustsson, M.T.; Lauko, A.; Sravya, P.; Røsland, G.V.; et al. GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat. Cancer 2023, 4, 648–664. [Google Scholar] [CrossRef]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef]
- Kim, D.; Ko, H.Y.; Chung, J.I.; Park, Y.M.; Lee, S.; Kim, S.Y.; Kim, J.; Chun, J.H.; Han, K.S.; Lee, M.; et al. Visualizing Cancer-Originating Acetate Uptake Through MCT1 in Reactive Astrocytes in the Glioblastoma Tumor Microenvironment. Neuro Oncol. 2023, 26, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jiang, Y.; Zhang, H.; Zhou, J.; Chen, L.; Li, H.; Xu, J.; Zhang, G.; Jing, Z. The SRSF1/circATP5B/miR-185-5p/HOXB5 feedback loop regulates the proliferation of glioma stem cells via the IL6-mediated JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2021, 40, 134. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Y.; O’Brien, J.A.; Franco-Barraza, J.; Qi, X.; Yuan, H.; Jin, W.; Zhang, J.; Gu, C.; Zhao, Z.; et al. Necroptotic astrocytes contribute to maintaining stemness of disseminated medulloblastoma through CCL2 secretion. Neuro Oncol. 2020, 22, 625–638. [Google Scholar] [CrossRef]
- Jin, P.; Shin, S.H.; Chun, Y.S.; Shin, H.W.; Shin, Y.J.; Lee, Y.; Kim, D.; Nam, D.H.; Park, J.W. Astrocyte-derived CCL20 reinforces HIF-1-mediated hypoxic responses in glioblastoma by stimulating the CCR6-NF-κB signaling pathway. Oncogene 2018, 37, 3070–3087. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, J.; Geng, X.; Li, S.; Zou, Y.; Li, Y.; Jing, C.; Yu, H. The Reactive Astrocytes After Surgical Brain Injury Potentiates the Migration, Invasion, and Angiogenesis of C6 Glioma. World Neurosurg. 2022, 168, e595–e606. [Google Scholar] [CrossRef]
- Perelroizen, R.; Philosof, B.; Budick-Harmelin, N.; Chernobylsky, T.; Ron, A.; Katzir, R.; Shimon, D.; Tessler, A.; Adir, O.; Gaoni-Yogev, A.; et al. Astrocyte immunometabolic regulation of the tumour microenvironment drives glioblastoma pathogenicity. Brain 2022, 145, 3288–3307. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef]
- Chen, W.; Wang, D.; Du, X.; He, Y.; Chen, S.; Shao, Q.; Ma, C.; Huang, B.; Chen, A.; Zhao, P.; et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med. Oncol. 2015, 32, 43. [Google Scholar] [CrossRef]
- Berg, T.J.; Marques, C.; Pantazopoulou, V.; Johansson, E.; von Stedingk, K.; Lindgren, D.; Jeannot, P.; Pietras, E.J.; Bergström, T.; Swartling, F.J.; et al. The Irradiated Brain Microenvironment Supports Glioma Stemness and Survival via Astrocyte-Derived Transglutaminase 2. Cancer Res. 2021, 81, 2101–2115. [Google Scholar] [CrossRef]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Sananikone, E.; Kanji, S.; Tomimatsu, N.; Di Cristofaro, L.F.M.; Kollipara, R.K.; Saha, D.; Floyd, J.R.; Sung, P.; Hromas, R.; Burns, T.C.; et al. Elimination of Radiation-Induced Senescence in the Brain Tumor Microenvironment Attenuates Glioblastoma Recurrence. Cancer Res. 2021, 81, 5935–5947. [Google Scholar] [CrossRef]
- Tanay, A.; Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 2017, 541, 331–338. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Bardehle, S.; Krüger, M.; Buggenthin, F.; Schwausch, J.; Ninkovic, J.; Clevers, H.; Snippert, H.J.; Theis, F.J.; Meyer-Luehmann, M.; Bechmann, I.; et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 2013, 16, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, G.; Yang, L.; Li, Z.; Zhang, Z.; Xu, Z.; Cai, Y.; Du, H.; Su, Z.; Wang, Z.; et al. Decoding Cortical Glial Cell Development. Neurosci. Bull. 2021, 37, 440–460. [Google Scholar] [CrossRef]
- Bachoo, R.M.; Maher, E.A.; Ligon, K.L.; Sharpless, N.E.; Chan, S.S.; You, M.J.; Tang, Y.; DeFrances, J.; Stover, E.; Weissleder, R.; et al. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Endersby, R.; Zhu, X.; Hay, N.; Ellison, D.W.; Baker, S.J. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res. 2011, 71, 4106–4116. [Google Scholar] [CrossRef]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef]
- Chow, L.M.; Endersby, R.; Zhu, X.; Rankin, S.; Qu, C.; Zhang, J.; Broniscer, A.; Ellison, D.W.; Baker, S.J. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 2011, 19, 305–316. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Li, Y.; Laterra, J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res. 2012, 72, 576–580. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Ventura, P.B.; Jiang, Y.; Rodriguez, F.J.; Wang, L.; Perry, J.S.A.; Yang, Y.; Wahl, K.; Crittenden, R.B.; Bennett, M.L.; et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 2020, 180, 502–520.e519. [Google Scholar] [CrossRef] [PubMed]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Jacque, C.M.; Vinner, C.; Kujas, M.; Raoul, M.; Racadot, J.; Baumann, N.A. Determination of glial fibrillary acidic protein (GFAP) in human brain tumors. J. Neurol. Sci. 1978, 35, 147–155. [Google Scholar] [CrossRef]
- Gagliano, N.; Costa, F.; Cossetti, C.; Pettinari, L.; Bassi, R.; Chiriva-Internati, M.; Cobos, E.; Gioia, M.; Pluchino, S. Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol. Rep. 2009, 22, 1349–1356. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Ding, S.; Wang, C.; Wang, W.; Yu, H.; Chen, B.; Liu, L.; Zhang, M.; Lang, Y. Autocrine S100B in astrocytes promotes VEGF-dependent inflammation and oxidative stress and causes impaired neuroprotection. Cell Biol. Toxicol. 2023, 39, 1–25. [Google Scholar] [CrossRef]
- Langeh, U.; Singh, S. Targeting S100B Protein as a Surrogate Biomarker and its Role in Various Neurological Disorders. Curr. Neuropharmacol. 2021, 19, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. S100B Protein Regulates Astrocyte Shape and Migration via Interaction with Src Kinase: Implications for Astrocyte Development, Activation, and Tumor Growth. J. Biol. Chem. 2009, 284, 8797–8811. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Zhang, I.Y.; Chen, X.; Da Fonseca, A.; Wu, S.; Ren, H.; Badie, S.; Sadeghi, S.; Ouyang, M.; et al. S100B promotes glioma growth through chemoattraction of myeloid-derived macrophages. Clin. Cancer Res. 2013, 19, 3764–3775. [Google Scholar] [CrossRef] [PubMed]
- Al-Dalahmah, O.; Argenziano, M.G.; Kannan, A.; Mahajan, A.; Furnari, J.; Paryani, F.; Boyett, D.; Save, A.; Humala, N.; Khan, F.; et al. Re-convolving the compositional landscape of primary and recurrent glioblastoma reveals prognostic and targetable tissue states. Nat. Commun. 2023, 14, 2586. [Google Scholar] [CrossRef]
- Boison, D.; Steinhäuser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [CrossRef]
- Gosejacob, D.; Dublin, P.; Bedner, P.; Hüttmann, K.; Zhang, J.; Tress, O.; Willecke, K.; Pfrieger, F.; Steinhäuser, C.; Theis, M. Role of astroglial connexin30 in hippocampal gap junction coupling. Glia 2011, 59, 511–519. [Google Scholar] [CrossRef]
- Griemsmann, S.; Höft, S.P.; Bedner, P.; Zhang, J.; von Staden, E.; Beinhauer, A.; Degen, J.; Dublin, P.; Cope, D.W.; Richter, N.; et al. Characterization of Panglial Gap Junction Networks in the Thalamus, Neocortex, and Hippocampus Reveals a Unique Population of Glial Cells. Cereb. Cortex 2015, 25, 3420–3433. [Google Scholar] [CrossRef]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Bedner, P.; Steinhäuser, C. Role of Impaired Astrocyte Gap Junction Coupling in Epileptogenesis. Cells 2023, 12, 1669. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Borrachero-Conejo, A.I.; Adams, W.R.; Saracino, E.; Mola, M.G.; Wang, M.; Posati, T.; Formaggio, F.; De Bellis, M.; Frigeri, A.; Caprini, M.; et al. Stimulation of water and calcium dynamics in astrocytes with pulsed infrared light. FASEB J. 2020, 34, 6539–6553. [Google Scholar] [CrossRef]
- Zhang, W.; Couldwell, W.T.; Simard, M.F.; Song, H.; Lin, J.H.; Nedergaard, M. Direct gap junction communication between malignant glioma cells and astrocytes. Cancer Res. 1999, 59, 1994–2003. [Google Scholar] [PubMed]
- Uzu, M.; Sin, W.C.; Shimizu, A.; Sato, H. Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1159. [Google Scholar] [CrossRef]
- He, Q.; Dent, E.W.; Meiri, K.F. Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J. Neurosci. 1997, 17, 3515–3524. [Google Scholar] [CrossRef]
- Nguyen, L.; He, Q.; Meiri, K.F. Regulation of GAP-43 at serine 41 acts as a switch to modulate both intrinsic and extrinsic behaviors of growing neurons, via altered membrane distribution. Mol. Cell Neurosci. 2009, 41, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Huang, Z.Y.; Wu, Y.; Burke, S.P.; Gutmann, D.H. The 43000 growth-associated protein functions as a negative growth regulator in glioma. Cancer Res 2003, 63, 2933–2939. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol. 2015, 37, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Burbach, G.J.; Hellweg, R.; Haas, C.A.; Del Turco, D.; Deicke, U.; Abramowski, D.; Jucker, M.; Staufenbiel, M.; Deller, T. Induction of brain-derived neurotrophic factor in plaque-associated glial cells of aged APP23 transgenic mice. J. Neurosci. 2004, 24, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Froger, N.; Ezan, P.; Jiang, J.X.; Bennett, M.V.; Naus, C.C.; Giaume, C.; Sáez, J.C. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 2011, 118, 826–840. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Feng, A.; Rock, R.B. Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem. Res. 2013, 38, 2148–2159. [Google Scholar] [CrossRef]
- Placone, A.L.; Quiñones-Hinojosa, A.; Searson, P.C. The role of astrocytes in the progression of brain cancer: Complicating the picture of the tumor microenvironment. Tumour Biol. 2016, 37, 61–69. [Google Scholar] [CrossRef]
- Zamykal, M.; Martens, T.; Matschke, J.; Günther, H.S.; Kathagen, A.; Schulte, A.; Peters, R.; Westphal, M.; Lamszus, K. Inhibition of intracerebral glioblastoma growth by targeting the insulin-like growth factor 1 receptor involves different context-dependent mechanisms. Neuro Oncol. 2015, 17, 1076–1085. [Google Scholar] [CrossRef]
- Chen, P.; Wang, W.; Liu, R.; Lyu, J.; Zhang, L.; Li, B.; Qiu, B.; Tian, A.; Jiang, W.; Ying, H.; et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature 2022, 606, 550–556. [Google Scholar] [CrossRef]
- Osuka, S.; Zhu, D.; Zhang, Z.; Li, C.; Stackhouse, C.T.; Sampetrean, O.; Olson, J.J.; Gillespie, G.Y.; Saya, H.; Willey, C.D.; et al. N-cadherin upregulation mediates adaptive radioresistance in glioblastoma. J. Clin. Investig. 2021, 131, e136098. [Google Scholar] [CrossRef]
- Yu, T.; Wang, X.; Zhi, T.; Zhang, J.; Wang, Y.; Nie, E.; Zhou, F.; You, Y.; Liu, N. Delivery of MGMT mRNA to glioma cells by reactive astrocyte-derived exosomes confers a temozolomide resistance phenotype. Cancer Lett. 2018, 433, 210–220. [Google Scholar] [CrossRef]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.B.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 22, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xiong, M.; Chen, R.; Ma, Y.; Corman, C.; Maricos, M.; Kindler, U.; Semtner, M.; Chen, Y.H.; Dahiya, S.; et al. Athymic mice reveal a requirement for T-cell-microglia interactions in establishing a microenvironment supportive of Nf1 low-grade glioma growth. Genes. Dev. 2018, 32, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e1514. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Dranoff, G.; Elion, G.B.; Friedman, H.S.; Campbell, G.L.; Bigner, D.D. Influence of glutamine on the growth of human glioma and medulloblastoma in culture. Cancer Res. 1985, 45, 4077–4081. [Google Scholar] [PubMed]
- Xu, J.; Ji, T.; Li, G.; Zhang, H.; Zheng, Y.; Li, M.; Ma, J.; Li, Y.; Chi, G. Lactate attenuates astrocytic inflammation by inhibiting ubiquitination and degradation of NDRG2 under oxygen-glucose deprivation conditions. J. Neuroinflamm. 2022, 19, 314. [Google Scholar] [CrossRef]
- Xian, P.; Hei, Y.; Wang, R.; Wang, T.; Yang, J.; Li, J.; Di, Z.; Liu, Z.; Baskys, A.; Liu, W.; et al. Mesenchymal stem cell-derived exosomes as a nanotherapeutic agent for amelioration of inflammation-induced astrocyte alterations in mice. Theranostics 2019, 9, 5956–5975. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Jahan, N.; Hannila, S.S. Transforming growth factor β-induced expression of chondroitin sulfate proteoglycans is mediated through non-Smad signaling pathways. Exp. Neurol. 2015, 263, 372–384. [Google Scholar] [CrossRef]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Field, C.S.; Baixauli, F.; Kyle, R.L.; Puleston, D.J.; Cameron, A.M.; Sanin, D.E.; Hippen, K.L.; Loschi, M.; Thangavelu, G.; Corrado, M.; et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2020, 31, 422–437.e425. [Google Scholar] [CrossRef]
- Kubota, Y.; Suda, T. Feedback mechanism between blood vessels and astrocytes in retinal vascular development. Trends Cardiovasc. Med. 2009, 19, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Liu, Q.; Lee, H.S.; Hossain, M.G.; Lacy-Hulbert, A.; McCarty, J.H. The astrocyte-expressed integrin αvβ8 governs blood vessel sprouting in the developing retina. Development 2011, 138, 5157–5166. [Google Scholar] [CrossRef]
- O’Sullivan, M.L.; Puñal, V.M.; Kerstein, P.C.; Brzezinski, J.A.t.; Glaser, T.; Wright, K.M.; Kay, J.N. Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia 2017, 65, 1697–1716. [Google Scholar] [CrossRef]
- Puebla, M.; Tapia, P.J.; Espinoza, H. Key Role of Astrocytes in Postnatal Brain and Retinal Angiogenesis. Int. J. Mol. Sci. 2022, 23, 2646. [Google Scholar] [CrossRef]
- Li, X.; Xue, Y.; Liu, X.; Zheng, J.; Shen, S.; Yang, C.; Chen, J.; Li, Z.; Liu, L.; Ma, J.; et al. ZRANB2/SNHG20/FOXK1 Axis regulates Vasculogenic mimicry formation in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 68. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, X.; Zhang, Y.; Mo, Y.; Sun, X.; Shu, L.; Ke, Y. Paradoxical role of β8 integrin on angiogenesis and vasculogenic mimicry in glioblastoma. Cell Death Dis. 2022, 13, 536. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic mimicry in carcinogenesis and clinical applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef]
- Bozoyan, L.; Khlghatyan, J.; Saghatelyan, A. Astrocytes control the development of the migration-promoting vasculature scaffold in the postnatal brain via VEGF signaling. J. Neurosci. 2012, 32, 1687–1704. [Google Scholar] [CrossRef]
- Gómez-Pinilla, F.; Lee, J.W.; Cotman, C.W. Distribution of basic fibroblast growth factor in the developing rat brain. Neuroscience 1994, 61, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Proia, P.; Schiera, G.; Mineo, M.; Ingrassia, A.M.; Santoro, G.; Savettieri, G.; Di Liegro, I. Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 2008, 21, 63–67. [Google Scholar]
- Acker, T.; Beck, H.; Plate, K.H. Cell type specific expression of vascular endothelial growth factor and angiopoietin-1 and -2 suggests an important role of astrocytes in cerebellar vascularization. Mech. Dev. 2001, 108, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.; Williams, J.; Nathans, J. Roles of HIFs and VEGF in angiogenesis in the retina and brain. J. Clin. Investig. 2019, 129, 3807–3820. [Google Scholar] [CrossRef]
- Zong, X.; Li, Y.; Liu, C.; Qi, W.; Han, D.; Tucker, L.; Dong, Y.; Hu, S.; Yan, X.; Zhang, Q. Theta-burst transcranial magnetic stimulation promotes stroke recovery by vascular protection and neovascularization. Theranostics 2020, 10, 12090–12110. [Google Scholar] [CrossRef] [PubMed]
- Pantazopoulou, V.; Jeannot, P.; Rosberg, R.; Berg, T.J.; Pietras, A. Hypoxia-Induced Reactivity of Tumor-Associated Astrocytes Affects Glioma Cell Properties. Cells 2021, 10, 613. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Pham, C.; Komaki, Y.; Deàs-Just, A.; Le Gac, B.; Mouffle, C.; Franco, C.; Chaperon, A.; Vialou, V.; Tsurugizawa, T.; Cauli, B.; et al. Astrocyte aquaporin mediates a tonic water efflux maintaining brain homeostasis. elife 2024, 13, RP95873. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Törnroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2022, 145, 64–75. [Google Scholar] [CrossRef]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018, 13, 379–394. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Si, X.; Dai, S.; Fang, Y.; Tang, J.; Wang, Z.; Li, Y.; Song, Z.; Chen, Y.; Liu, Y.; Zhao, G.; et al. Matrix metalloproteinase-9 inhibition prevents aquaporin-4 depolarization-mediated glymphatic dysfunction in Parkinson’s disease. J. Adv. Res. 2024, 56, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Reeves, B.C.; Karimy, J.K.; Kundishora, A.J.; Mestre, H.; Cerci, H.M.; Matouk, C.; Alper, S.L.; Lundgaard, I.; Nedergaard, M.; Kahle, K.T. Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol. Med. 2020, 26, 285–295. [Google Scholar] [CrossRef]
- Sun, C.; Lin, L.; Yin, L.; Hao, X.; Tian, J.; Zhang, X.; Ren, Y.; Li, C.; Yang, Y. Acutely Inhibiting AQP4 With TGN-020 Improves Functional Outcome by Attenuating Edema and Peri-Infarct Astrogliosis After Cerebral Ischemia. Front. Immunol. 2022, 13, 870029. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Krishna, S.; Bell, B.A. Aquaporin-4 expression is increased in oedematous human brain tumours. J. Neurol. Neurosurg. Psychiatry 2002, 72, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e1016. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773.e758. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Fathali, N.; Doyle, K.P.; Williams, A.M.; Han, J.; Buckwalter, M.S. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 2014, 62, 1227–1240. [Google Scholar] [CrossRef]
- Jayaram, M.A.; Phillips, J.J. Role of the Microenvironment in Glioma Pathogenesis. Annu. Rev. Pathol. 2024, 19, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Ribot, J.; Breton, R.; Calvo, C.F.; Moulard, J.; Ezan, P.; Zapata, J.; Samama, K.; Moreau, M.; Bemelmans, A.P.; Sabatet, V.; et al. Astrocytes close the mouse critical period for visual plasticity. Science 2021, 373, 77–81. [Google Scholar] [CrossRef]
- Haylock-Jacobs, S.; Keough, M.B.; Lau, L.; Yong, V.W. Chondroitin sulphate proteoglycans: Extracellular matrix proteins that regulate immunity of the central nervous system. Autoimmun. Rev. 2011, 10, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Saito, A.; Kamikawa, Y.; Ito, T.; Matsuhisa, K.; Kaneko, M.; Okamoto, T.; Yoshimaru, T.; Matsushita, Y.; Katagiri, T.; Imaizumi, K. p53-independent tumor suppression by cell-cycle arrest via CREB/ATF transcription factor OASIS. Cell Rep. 2023, 42, 112479. [Google Scholar] [CrossRef]
- Chen, K.Z.; Liu, S.X.; Li, Y.W.; He, T.; Zhao, J.; Wang, T.; Qiu, X.X.; Wu, H.F. Vimentin as a potential target for diverse nervous system diseases. Neural Regen. Res. 2023, 18, 969–975. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of vimentin in health and disease. Genes. Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]
- Lu, P.; Wang, Y.; Liu, X.; Wang, H.; Zhang, X.; Wang, K.; Wang, Q.; Hu, R. Malignant gliomas induce and exploit astrocytic mesenchymal-like transition by activating canonical Wnt/β-catenin signaling. Med. Oncol. 2016, 33, 66. [Google Scholar] [CrossRef]
- Stupp, R.; Tonn, J.C.; Brada, M.; Pentheroudakis, G. High-grade malignant glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. 5), v190–v193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, R.; Yu, Y.; Liu, J.; Luo, T.; Fan, F. Glioblastoma Treatment Modalities besides Surgery. J. Cancer 2019, 10, 4793–4806. [Google Scholar] [CrossRef]
- Niklasson, M.; Bergström, T.; Jarvius, M.; Sundström, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal transition and increased therapy resistance of glioblastoma cells is related to astrocyte reactivity. J. Pathol. 2019, 249, 295–307. [Google Scholar] [CrossRef]
- Alberghina, C.; Torrisi, F.; D’Aprile, S.; Longhitano, L.; Giallongo, S.; Scandura, G.; Mannino, G.; Mele, S.; Sabini, M.G.; Cammarata, F.P.; et al. Microglia and glioblastoma heterocellular interplay sustains tumour growth and proliferation as an off-target effect of radiotherapy. Cell Prolif. 2024, 57, e13606. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Rose, I.V.L.; Kim, H.; Xia, W.; Romero-Fernandez, W.; Rooney, B.; Koontz, M.; Li, E.; Ao, Y.; Wang, S.; et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat. Neurosci. 2022, 25, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Nakano-Kobayashi, A.; Canela, A.; Yoshihara, T.; Hagiwara, M. Astrocyte-targeting therapy rescues cognitive impairment caused by neuroinflammation via the Nrf2 pathway. Proc. Natl. Acad. Sci. USA 2023, 120, e2303809120. [Google Scholar] [CrossRef]
- Jung, B.K.; Park, Y.; Yoon, B.; Bae, J.S.; Han, S.W.; Heo, J.E.; Kim, D.E.; Ryu, K.Y. Reduced secretion of LCN2 (lipocalin 2) from reactive astrocytes through autophagic and proteasomal regulation alleviates inflammatory stress and neuronal damage. Autophagy 2023, 19, 2296–2317. [Google Scholar] [CrossRef]
- Liu, H.; Wu, X.; Luo, J.; Wang, X.; Guo, H.; Feng, D.; Zhao, L.; Bai, H.; Song, M.; Liu, X.; et al. Pterostilbene Attenuates Astrocytic Inflammation and Neuronal Oxidative Injury After Ischemia-Reperfusion by Inhibiting NF-κB Phosphorylation. Front. Immunol. 2019, 10, 2408. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Li, X.; Zhang, Y.; Rasouli, J.; Casella, G.; Boehm, A.; Hwang, D.; Ishikawa, L.L.; Thome, R.; et al. SIRT1 inactivation switches reactive astrocytes to an antiinflammatory phenotype in CNS autoimmunity. J. Clin. Investig. 2022, 132, e151803. [Google Scholar] [CrossRef]
- Zhong, Y.; Geng, F.; Mazik, L.; Yin, X.; Becker, A.P.; Mohammed, S.; Su, H.; Xing, E.; Kou, Y.; Chiang, C.Y.; et al. Combinatorial targeting of glutamine metabolism and lysosomal-based lipid metabolism effectively suppresses glioblastoma. Cell Rep. Med. 2024, 5, 101706. [Google Scholar] [CrossRef]
- Zeng, L.; Zheng, W.; Liu, X.; Zhou, Y.; Jin, X.; Xiao, Y.; Bai, Y.; Pan, Y.; Zhang, J.; Shao, C. SDC1-TGM2-FLOT1-BHMT complex determines radiosensitivity of glioblastoma by influencing the fusion of autophagosomes with lysosomes. Theranostics 2023, 13, 3725–3743. [Google Scholar] [CrossRef]
- Sandhu, M.R.S.; Gruenbaum, B.F.; Gruenbaum, S.E.; Dhaher, R.; Deshpande, K.; Funaro, M.C.; Lee, T.W.; Zaveri, H.P.; Eid, T. Astroglial Glutamine Synthetase and the Pathogenesis of Mesial Temporal Lobe Epilepsy. Front. Neurol. 2021, 12, 665334. [Google Scholar] [CrossRef]
- Du, L.; Xing, Z.; Tao, B.; Li, T.; Yang, D.; Li, W.; Zheng, Y.; Kuang, C.; Yang, Q. Both IDO1 and TDO contribute to the malignancy of gliomas via the Kyn-AhR-AQP4 signaling pathway. Signal Transduct. Target. Ther. 2020, 5, 10. [Google Scholar] [CrossRef]
- Simone, L.; Pisani, F.; Mola, M.G.; De Bellis, M.; Merla, G.; Micale, L.; Frigeri, A.; Vescovi, A.L.; Svelto, M.; Nicchia, G.P. AQP4 Aggregation State Is a Determinant for Glioma Cell Fate. Cancer Res. 2019, 79, 2182–2194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, W.; Zhang, X.; Huang, B.; Chen, A.; He, Y.; Wang, J.; Li, X. Galunisertib inhibits glioma vasculogenic mimicry formation induced by astrocytes. Sci. Rep. 2016, 6, 23056. [Google Scholar] [CrossRef]
- Li, H.; Zhu, J.; Liu, X.; Liu, L.; Huang, S.; Wu, A.; Xu, Z.; Zhang, X.; Li, Z.; Ni, F.; et al. Glioma stem cell-derived exosomes induce the transformation of astrocytes via the miR-3065-5p/DLG2 signaling axis. Glia 2024, 72, 857–871. [Google Scholar] [CrossRef]
- Rosito, M.; Maqbool, J.; Reccagni, A.; Mangano, M.; D’Andrea, T.; Rinaldi, A.; Peruzzi, G.; Silvestri, B.; Rosa, A.; Trettel, F.; et al. Ketogenic diet induces an inflammatory reactive astrocytes phenotype reducing glioma growth. Cell Mol. Life Sci. 2025, 82, 73. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, W.; Gou, C.; Wang, X.; Wang, X.; Shao, X.; Chen, X.; Chen, Z. AS1411 binds to nucleolin via its parallel structure and disrupts the exos-miRNA-27a-mediated reciprocal activation loop between glioma and astrocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167211. [Google Scholar] [CrossRef]
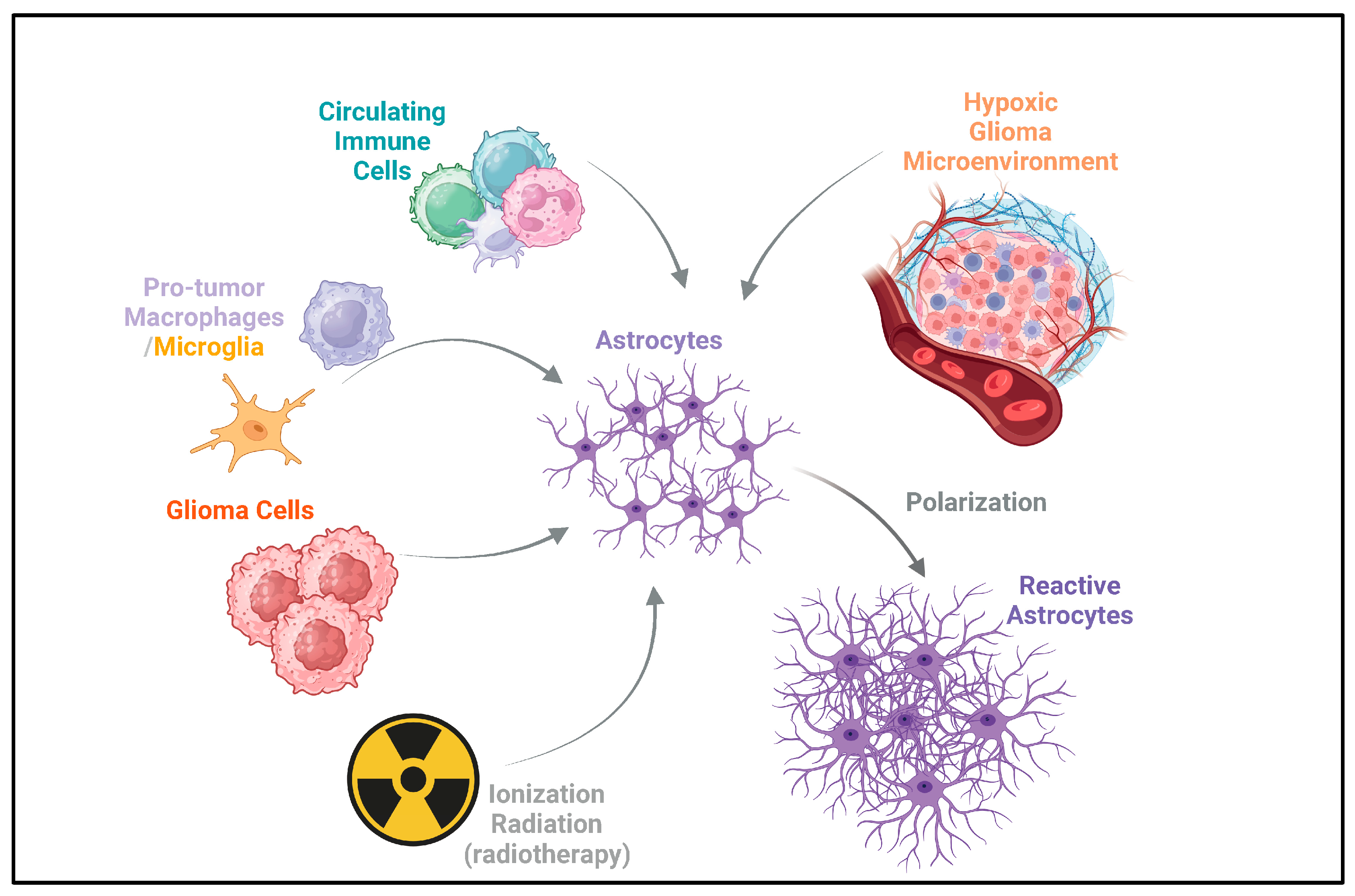
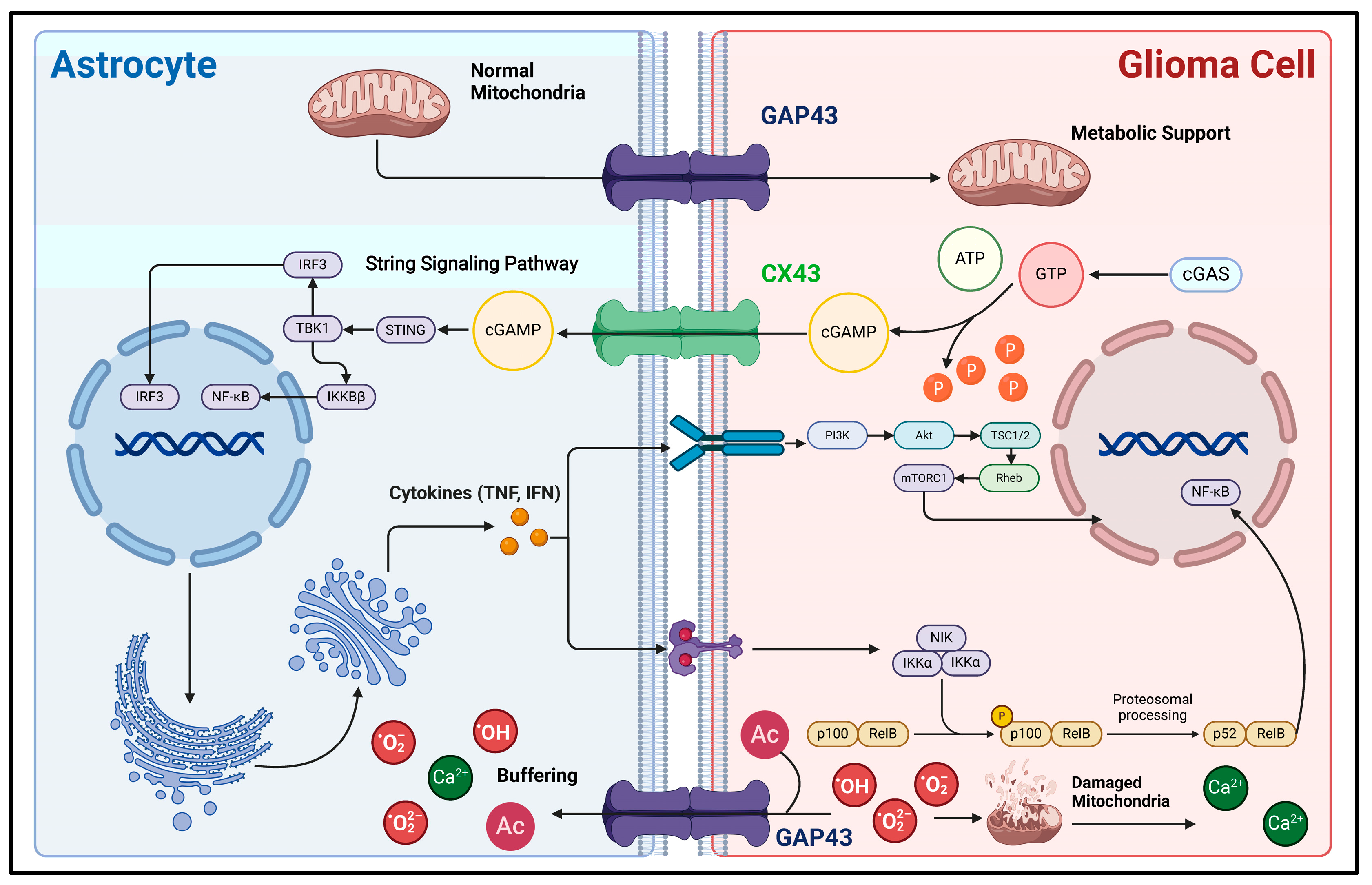
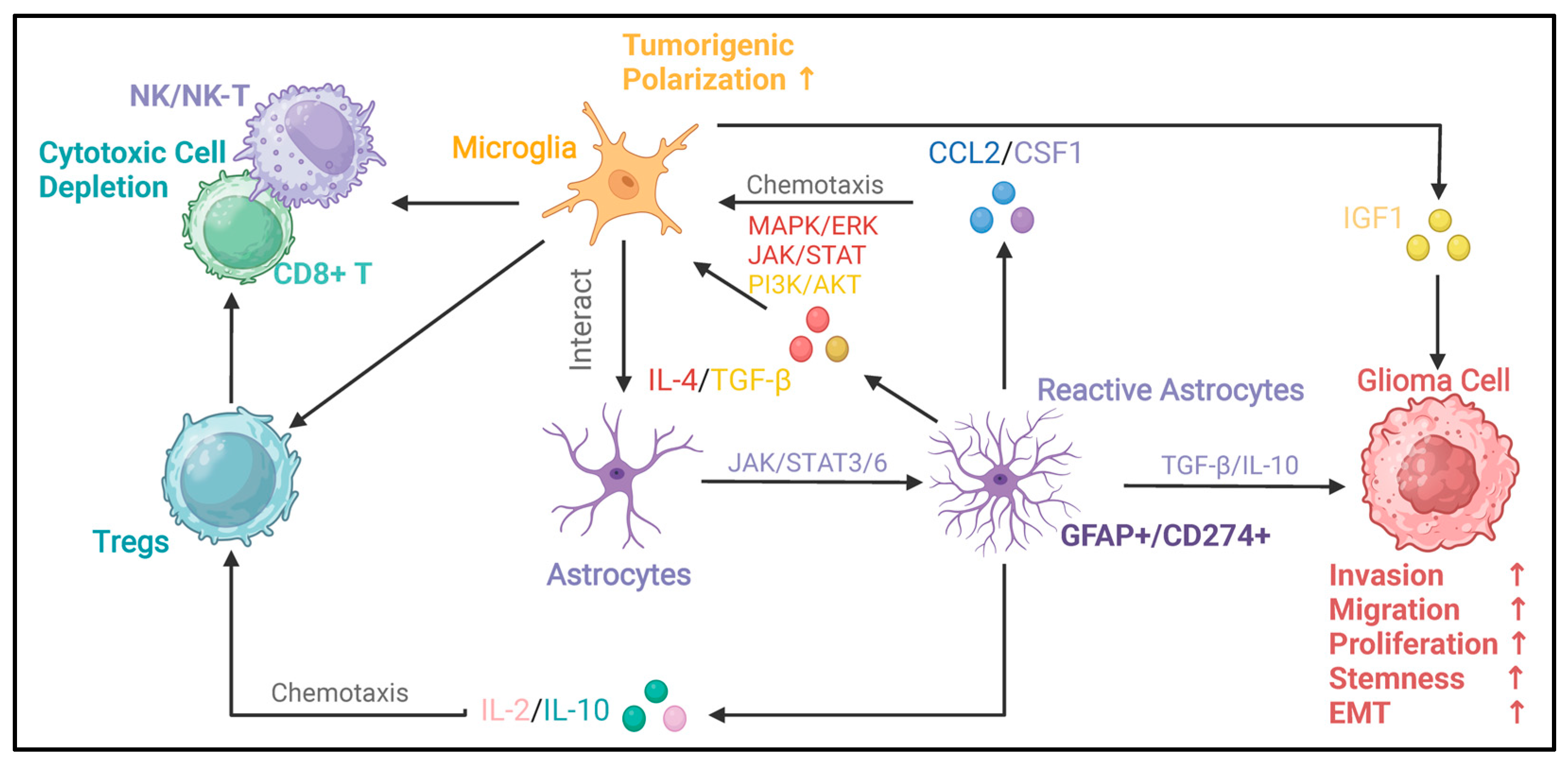
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).