
High Tumor Mutation Burden (TMB) and a Novel Somatic Mutation in the TREX1 Gene in a Patient with Aggressive and Refractory High-Grade B-Cell Lymphoma: A Case Report

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Case Report
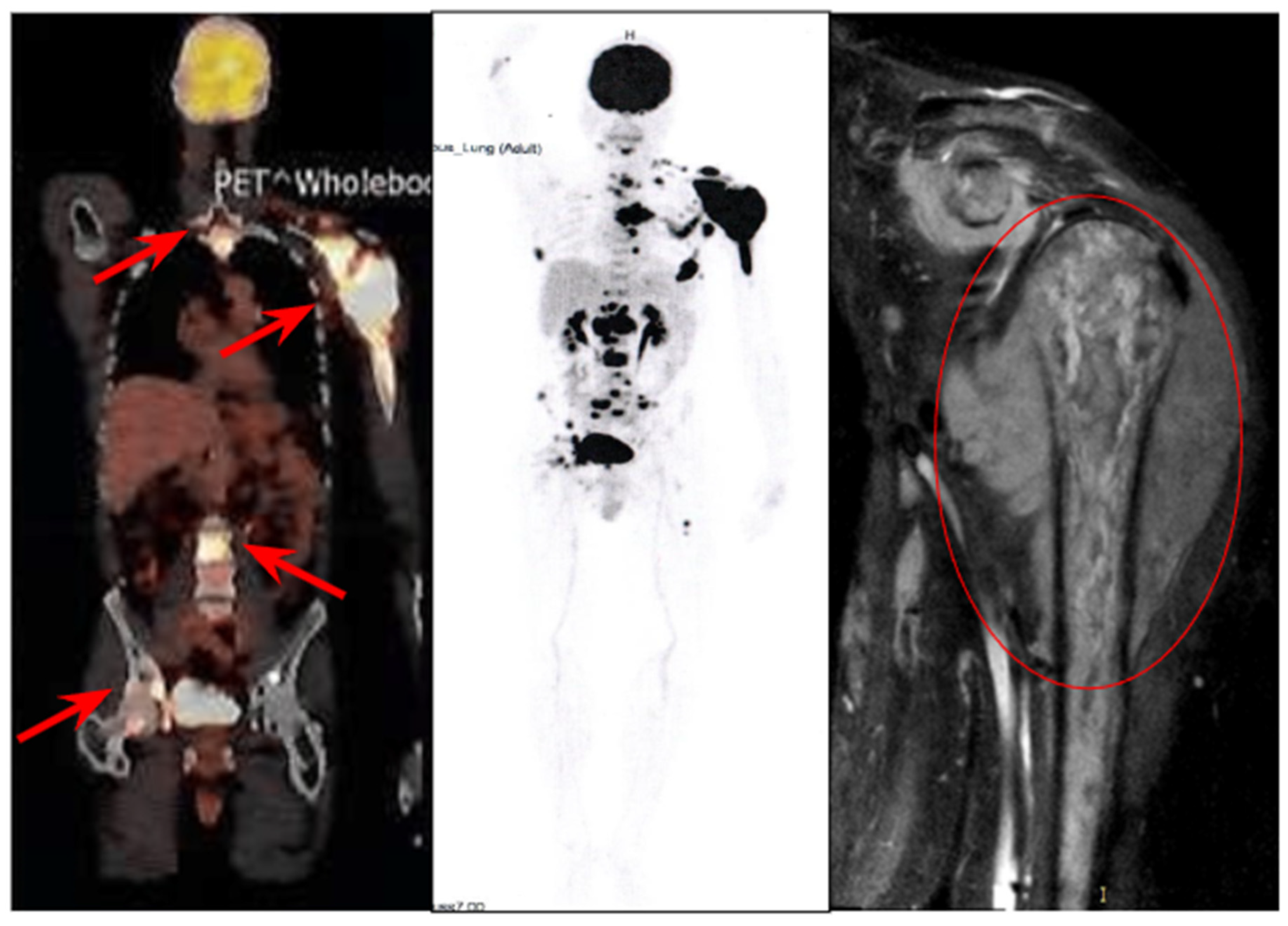
2.1. History of the Disease and Treatment
2.2. Retrospective Assessment of the Tumor Molecular Profile
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842. [Google Scholar] [PubMed]
- Dunleavy, K. Double-Hit Lymphoma: Optimizing Therapy. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 157. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375. [Google Scholar]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [PubMed]
- Davies, A.J. The High-Grade B-Cell Lymphomas: Double Hit and More. Blood 2024, 144, 2583–2592. [Google Scholar] [PubMed]
- Moharana, L.; Dasappa, L.; Babu, S.; Lokesh, K.N.; Rudresh, A.; Rajeev, L.K.; Saldanha, S.; Sharma, K.; Jacob, L.A. Comparison Between CHOP and DAEPOCH with or Without Rituximab in Adult High Grade B Cell Lymphoma, Not Otherwise Specified; A Retrospective Study From a Tertiary Cancer Hospital in South India. Indian J. Hematol. Blood Transfus. 2021, 38, 15–23. [Google Scholar]
- Chen, Y.; Cai, Q.; Chang, Y.; Zhang, M.; Li, Z. High-Intensity Chemotherapy Improved the Prognosis of Patients with High-Grade B-Cell Lymphoma. Front. Immunol. 2022, 13, 1047115. [Google Scholar]
- Olszewski, A.J.; Kurt, H.; Evens, A.M. Defining and Treating High-Grade B-Cell Lymphoma, NOS. Blood 2022, 140, 943–954. [Google Scholar]
- Karunakaran, P.; Selvarajan, G.; Kalaiyarasi, J.P.; Mehra, N.; Sundersingh, S.; Dhanushkodi, M.; Kesana, S.; Kannan, K.; Ganesan, T.S.; Radhakrishnan, V.; et al. Therapeutic Outcomes in High-Grade B-Cell Lymphoma, NOS: Retrospective Analysis. South Asian J. Cancer 2022, 11, 68–72. [Google Scholar]
- Brunner, J.R.; Altshuler, E.; Yang, L.-J. Analysis of the Diagnosis of Burkitt-like Lymphoma in a Patient with Atypical Cytogenetics and Molecular Markers. Cureus 2022, 14, e28295. [Google Scholar]
- Frontzek, F.; Lenz, G. Novel Insights into the Pathogenesis of Molecular Subtypes of Diffuse Large B-Cell Lymphoma and Their Clinical Implications. Expert Rev. Clin. Pharmacol. 2019, 12, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.W.L.; Weisenburger, D.D.; Greiner, T.C.; Piris, M.A.; Banham, A.H.; Delabie, J.; Braziel, R.M.; Geng, H.; Iqbal, J.; Lenz, G.; et al. A New Immunostain Algorithm Classifies Diffuse Large B-Cell Lymphoma into Molecular Subtypes with High Accuracy. Clin. Cancer Res. 2009, 15, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Boltežar, L.; Prevodnik, V.K.; Perme, M.P.; Gašljević, G.; Novaković, B.J. Comparison of the Algorithms Classifying the ABC and GCB Subtypes in Diffuse Large B-Cell Lymphoma. Oncol. Lett. 2018, 15, 6903–6912. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Fu, D.; Dong, L.; Zhang, M.-C.; Shi, Q.; Shi, Z.-Y.; Cheng, S.; Wang, L.; Xu, P.-P.; Zhao, W.-L. Simplified Algorithm for Genetic Subtyping in Diffuse Large B-Cell Lymphoma. Signal Transduct. Target. Ther. 2023, 8, 145. [Google Scholar] [CrossRef]
- Runge, H.F.P.; Lacy, S.; Barrans, S.; Beer, P.A.; Painter, D.; Smith, A.; Roman, E.; Burton, C.; Crouch, S.; Tooze, R.; et al. Application of the LymphGen Classification Tool to 928 Clinically and Genetically-Characterised Cases of Diffuse Large B Cell Lymphoma (DLBCL). Br. J. Haematol. 2021, 192, 216–220. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. JMD 2017, 19, 341–365. [Google Scholar]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSIsensor: Microsatellite Instability Detection Using Paired Tumor-Normal Sequence Data. Bioinformatics 2013, 30, 1015. [Google Scholar] [CrossRef]
- Dupain, C.; Gutman, T.; Girard, E.; Kamoun, C.; Marret, G.; Castel-Ajgal, Z.; Sablin, M.-P.; Neuzillet, C.; Borcoman, E.; Hescot, S.; et al. Tumor Mutational Burden Assessment and Standardized Bioinformatics Approach Using Custom NGS Panels in Clinical Routine. BMC Biol. 2024, 22, 43. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.2 List for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing: A Policy Statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2023, 25, 100866. [Google Scholar]
- Genomic Data Commons. Available online: https://docs.gdc.cancer.gov/Data/Introduction/ (accessed on 10 March 2025).
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- GnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 14 March 2025).
- Li, Q.; Ren, Z.; Cao, K.; Li, M.M.; Wang, K.; Zhou, Y. CancerVar: An Artificial Intelligence-Empowered Platform for Clinical Interpretation of Somatic Mutations in Cancer. Sci. Adv. 2022, 8, eabj1624. [Google Scholar] [PubMed]
- ClinVar ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 14 March 2025).
- COSMIC, the Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 12 January 2025).
- OncoKBTM—MSK’s Precision Oncology Knowledge Base. Available online: https://www.oncokb.org/ (accessed on 7 February 2025).
- CIViC—Clinical Interpretation of Variants in Cancer. Available online: https://civicdb.org/welcome (accessed on 7 February 2025).
- cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/ (accessed on 12 January 2025).
- PharmGKB. Available online: https://www.pharmgkb.org/ (accessed on 14 March 2025).
- PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 10 March 2025).
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 4685–4689. [Google Scholar]
- Agarwal, N.; Gupta, S.; Klempner, S.J.; Andrews, M.C.; Mahipal, A.; Subbiah, V.; Eskander, R.N.; Carbone, D.P.; Snider, J.; Bouzit, L.; et al. Tumor Mutational Burden (TMB) Measurement from an FDA-Approved Assay and Real-World Overall Survival (rwOS) on Single-Agent Immune Checkpoint Inhibitors (ICI) in over 8000 Patients across 24 Cancer Types. J. Clin. Oncol. 2023, 13, e010311. [Google Scholar]
- Griffin, R.; Wenzl, K.; Sarangi, V.; Rimsza, L.M.; King, R.; Feldman, A.L.; Maurer, M.J.; Nowakowski, G.S.; Link, B.K.; Habermann, T.M.; et al. Tumor Mutational Burden As a Prognostic Factor in Diffuse Large B-Cell Lymphoma. Blood 2023, 142, 1633. [Google Scholar]
- Epperla, N.; Zayac, A.S.; Landsburg, D.J.; Bock, A.M.; Nowakowski, G.S.; Ayers, E.C.; Girton, M.; Hu, M.; Beckman, A.; Li, S.; et al. High-Grade B-Cell Lymphoma, Not Otherwise Specified: CNS Involvement and Outcomes in a Multi-Institutional Series. Blood Adv. 2024, 8, 5355–5364. [Google Scholar]
- Li, S.; Qiu, L.; Xu, J.; Lin, P.; Ok, C.Y.; Tang, G.; McDonnell, T.J.; James You, M.; Khanlari, M.; Miranda, R.N.; et al. High-Grade B-Cell Lymphoma (HGBL)-NOS Is Clinicopathologically and Genetically More Similar to DLBCL/HGBL-DH than DLBCL. Leukemia 2022, 37, 422–432. [Google Scholar] [CrossRef]
- Dobashi, A.; Togashi, Y.; Tanaka, N.; Yokoyama, M.; Tsuyama, N.; Baba, S.; Mori, S.; Hatake, K.; Yamaguchi, T.; Noda, T.; et al. TP53 and OSBPL10 Alterations in Diffuse Large B-Cell Lymphoma: Prognostic Markers Identified via Exome Analysis of Cases with Extreme Prognosis. Oncotarget 2018, 9, 19555. [Google Scholar] [PubMed]
- He, M.; Liu, B.; Tang, G.; Jiao, L.; Liu, X.; Yin, S.; Wang, T.; Chen, J.; Gao, L.; Ni, X.; et al. B2M Mutation Paves the Way for Immune Tolerance in Pathogenesis of Epstein-Barr Virus Positive Diffuse Large B-Cell Lymphomas. J. Cancer 2022, 13, 3615. [Google Scholar]
- Liu, F.; Zhong, F.; Wu, H.; Che, K.; Shi, J.; Wu, N.; Fu, Y.; Wang, Y.; Hu, J.; Qian, X.; et al. Prevalence and Associations of Beta2-Microglobulin Mutations in MSI-H/dMMR Cancers. Oncology 2023, 28, e136. [Google Scholar]
- Benoit, A.; Abraham, M.J.; Li, S.; Kim, J.; Estrada-Tejedor, R.; Bakadlag, R.; Subramaniam, N.; Makhani, K.; Guilbert, C.; Tu, R.; et al. STAT6 Mutations Enriched at Diffuse Large B-Cell Lymphoma Relapse Reshape the Tumor Microenvironment. Int. J. Hematol. 2024, 119, 275. [Google Scholar] [PubMed]
- Tumor Immunotherapy Resistance: Revealing the Mechanism of PD-1/PD-L1-Mediated Tumor Immune Escape. Biomed. Pharmacother. 2024, 171, 116203.
- Song, T.L.; Nairismägi, M.-L.; Laurensia, Y.; Lim, J.-Q.; Tan, J.; Li, Z.-M.; Pang, W.-L.; Kizhakeyil, A.; Wijaya, G.-C.; Huang, D.-C.; et al. Oncogenic Activation of the STAT3 Pathway Drives PD-L1 Expression in Natural killer/T-Cell Lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar]
- Sabaawy, A.; Zeeshan, S. Targeting the Immune Microenvironment during Immunotherapy for Solid Tumors. Mol. Cell. Oncol. 2021, 8, 1994327. [Google Scholar]
- Morin, R.D.; Assouline, S.; Alcaide, M.; Mohajeri, A.; Johnston, R.L.; Chong, L.; Grewal, J.; Yu, S.; Fornika, D.; Bushell, K.; et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2290–2300. [Google Scholar]
- Świerczyńska, M.; Tronina, A.; Filipek, E. Aicardi–Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation. J. Pers. Med. 2023, 13, 1609. [Google Scholar] [CrossRef]
- VCV000282766.45—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/282766/ (accessed on 26 November 2024).
- Cuadrado, E.; Michailidou, I.; van Bodegraven, E.J.; Jansen, M.H.; Sluijs, J.A.; Geerts, D.; Couraud, P.O.; De Filippis, L.; Vescovi, A.L.; Kuijpers, T.W.; et al. Phenotypic Variation in Aicardi-Goutières Syndrome Explained by Cell-Specific IFN-Stimulated Gene Response and Cytokine Release. J. Immunol. 2015, 194, 3623–3633. [Google Scholar]
- Wang, C.-J.; Lam, W.; Bussom, S.; Chang, H.-M.; Cheng, Y.-C. TREX1 Acts in Degrading Damaged DNA from Drug-Treated Tumor Cells. DNA Repair. 2009, 8, 1179. [Google Scholar] [PubMed]
- Demaria, S.; Vanpouille-Box, C. TREX1 Is a Checkpoint for Innate Immune Sensing of DNA Damage That Fosters Cancer Immune Resistance. Emerg. Top. Life Sci. 2017, 1, 509–515. [Google Scholar]
- Hemphill, W.O.; Simpson, S.R.; Liu, M.; Salsbury, F.R.; Hollis, T.; Grayson, J.M.; Perrino, F.W. TREX1 as a Novel Immunotherapeutic Target. Front. Immunol. 2021, 12, 660184. [Google Scholar]
- Shim, A.; Luan, X.; Zhou, W.; Crow, Y.J.; Maciejowski, J. Mutations in the Non-Catalytic Polyproline Motif Destabilize TREX1 and Amplify cGAS-STING Signaling. Hum. Mol. Genet. 2024, 33, 1555–1566. [Google Scholar] [PubMed]
- Técher, H. T-Rex Escaped from the Cytosolic Park: Re-Thinking the Impact of TREX1 Exonuclease Deficiencies on Genomic Stability. BioEssays News Rev. Mol. Cell. Dev. Biol. 2024, 46, e2400066. [Google Scholar]
- Tani, T.; Mathsyaraja, H.; Campisi, M.; Li, Z.-H.; Haratani, K.; Fahey, C.G.; Ota, K.; Mahadevan, N.R.; Shi, Y.; Saito, S.; et al. TREX1 Inactivation Unleashes Cancer Cell STING-Interferon Signaling and Promotes Antitumor Immunity. Cancer Discov. 2024, 14, 752–765. [Google Scholar] [PubMed]
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The Crebbp Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B Cell Lymphoma. Cancer Discov. 2017, 7, 322. [Google Scholar]
- Thomas, A. CREBBP-Mutated Cancers HAT-Tricked. Sci. Transl. Med. 2015, 7, 317ec212. [Google Scholar]
- Liu, X.; Hu, X.; Zheng, Y. MA05.05 Analysis of CREBBP as a Potential Biomarker for Immune Checkpoint Therapy in Solid Tumors and Its Correlation with Immune Microenvironment. J. Thorac. Oncol. 2022, 17, S60. [Google Scholar]
- Chen, J.; Hu, X.; Zheng, Y.; Huang, M. Investigation of CREBBP Mutation and Correlation with Immunotherapy Biomarker in Chinese Bladder Cancer Patients. J. Clin. Oncol. 2022, 40, e16537. [Google Scholar]
- Su, W.; Feng, B.; Hu, L.; Guo, X.; Yu, M. MUC3A Promotes the Progression of Colorectal Cancer through the PI3K/Akt/mTOR Pathway. BMC Cancer 2022, 22, 602. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chin, C.R.; Ying, H.-Y.; Meydan, C.; Teater, M.R.; Xia, M.; Farinha, P.; Takata, K.; Chu, C.-S.; Jiang, Y.; et al. Loss of CREBBP and KMT2D Cooperate to Accelerate Lymphomagenesis and Shape the Lymphoma Immune Microenvironment. Nat. Commun. 2024, 15, 2879. [Google Scholar] [CrossRef]
- Budczies, J.; Kazdal, D.; Menzel, M.; Beck, S.; Kluck, K.; Altbürger, C.; Schwab, C.; Allgäuer, M.; Ahadova, A.; Kloor, M.; et al. Tumour Mutational Burden: Clinical Utility, Challenges and Emerging Improvements. Nat. Rev. Clin. Oncol. 2024, 21, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, L.; Zhang, S.; Tian, X.; De La Cruz, A.; George, A.; Arnoff, T.E.; El-Deiry, W.S. The Role of p53 in Anti-Tumor Immunity and Response to Immunotherapy. Front. Mol. Biosci. 2023, 10, 1148389. [Google Scholar] [CrossRef]
- van Kampen, F.; Clark, A.; Soul, J.; Kanhere, A.; Glenn, M.A.; Pettitt, A.R.; Kalakonda, N.; Slupsky, J.R. Deletion of 17p in Cancers: Guilt by (p53) Association. Oncogene 2025, 44, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Sweis, R.F.; Hodi, F.S.; Messersmith, W.A.; Andtbacka, R.H.I.; Ingham, M.; Lewis, N.; Chen, X.; Pelletier, M.; Chen, X.; et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 677–688. [Google Scholar] [CrossRef]
- Sallets, A.; Robinson, S.; Kardosh, A.; Levy, R. Enhancing Immunotherapy of STING Agonist for Lymphoma in Preclinical Models. Blood Adv. 2018, 2, 2230–2241. [Google Scholar] [CrossRef]
- Wang, B.; Yu, W.; Jiang, H.; Meng, X.; Tang, D.; Liu, D. Clinical Applications of STING Agonists in Cancer Immunotherapy: Current Progress and Future Prospects. Front. Immunol. 2024, 15, 1485546. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Marker | Meaning |
|---|---|
| CD20+ | Intense membrane expression |
| CD10+ | Intense cytoplasmic reaction |
| BCL6+ | Intense nuclear reaction |
| BCL2+ | Intense cytoplasmic reaction |
| Ki-67 | 90% |
| MUM1+ | Single small cells |
| CD23 | Reaction with CD23 antibody is questionable (negative/false negative?) |
| CD3+, CD5+, CD43+ | Few small lymphoid T-cells |
| c-MYC+ | 70–80% positive lymphoid infiltration cells (heterogeneous nuclear reaction) |
| TdT- | no TdT positive cells detected |
| GCB/non-GCB subtype | GCB subtype |
| Double-expressor (DEL) phenotype | Yes |
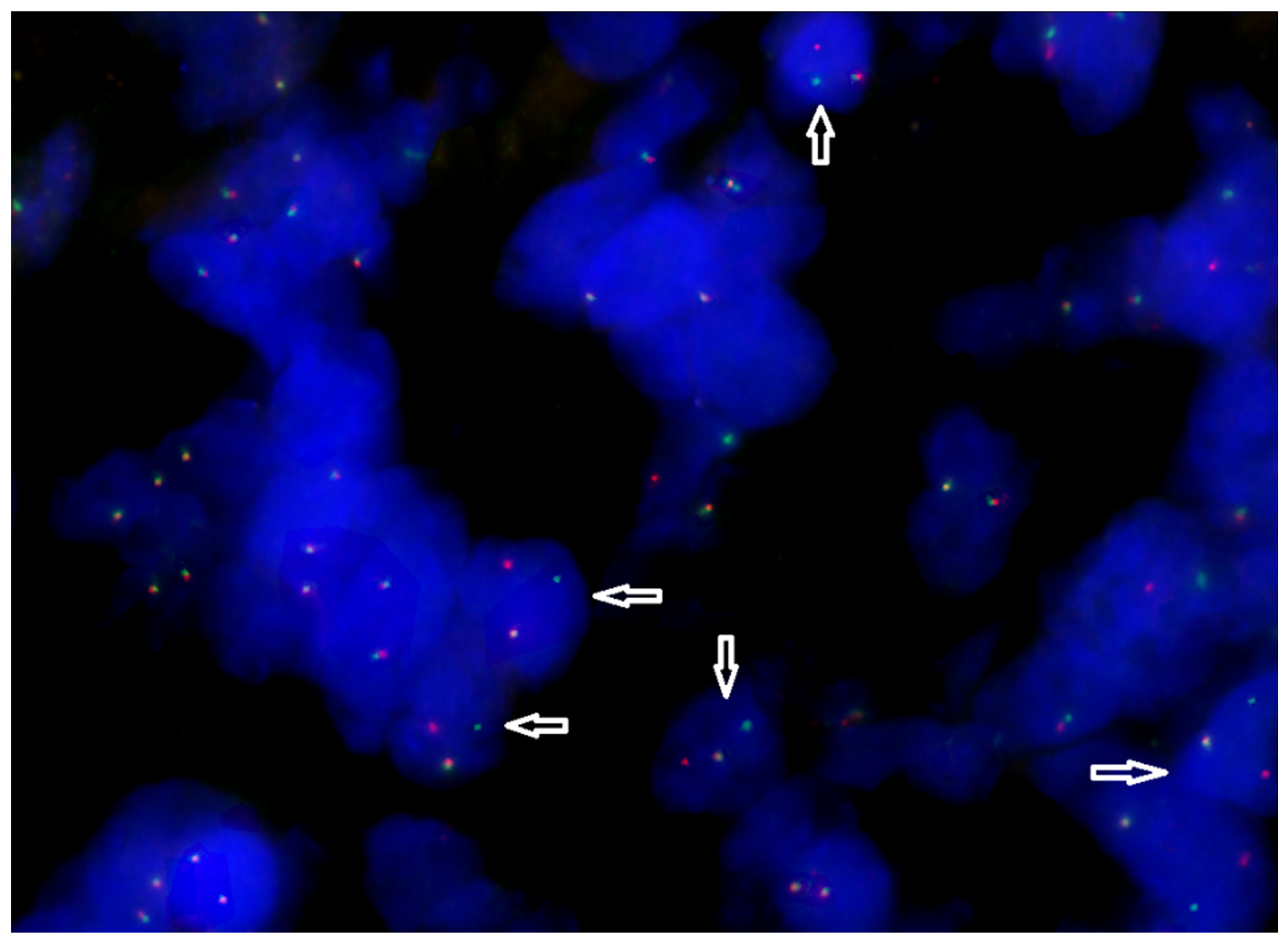
| Translocations (FISH) | BCL2 rearrangement and 17p13/TP53 deletion were detected; MYC, BCL6 rearrangements were not detected. |
| Karyotype (bone marrow) | 46, XY. Clonal chromosomal aberrations were not detected. |
| Date | Treatment | Result |
|---|---|---|
| 03.2020–07.2020 | R-CHOP №1, R-EPOCH №5 | Progression |
| 08.2020 | R-DHAP, methotrexate, lenalidomide, ibrutinib, venetoclax, nivolumab | Progression |
| 09.2020 | ifosfamide, dacarbazine, dexamethasone, mitoxantrone, obinutuzumab | Progression |
| 10.2020 | haploidentical hematopoietic stem cell transplantation (haplo-HSCT) | Progression. Death |
| Gene | Mutation | Classification | OncoKB [30] | COSMIC [29] | ClinVar [28] | AF (%)/Read Depths | Function | LOH |
|---|---|---|---|---|---|---|---|---|
| TP53 | NM_000546.6(TP53):c.712T>G (p.Cys238Gly) | Pathogenic | Likely Oncogenic Likely LOF Level 1 | COSV52711932 | Yes | 75%/16x | LOF | LOH |
| B2M | NM_004048:exon1:c.T35G:p.L12R | Probably pathogenic | Likely Oncogenic Unknown Biological Effect | COSV62563197 | - | 92,3%/46x | LOF | - |
| STAT6 | NM_003153:exon12:c.A1256G:p.D419G | Probably pathogenic | COSV55668829 | Yes | 66,7%/27x | GOF | - | |
| STAT3 | NM_213662:exon20:c.C1842G:p.S614R | Pathogenic | Likely Oncogenic Likely GOF Level 3 | COSV52888203 | - | 25%/52x | GOF | - |
| TREX1 | NM_033629.6(TREX1):c.144dup (p.Thr49fs) | Pathogenic | - | - | Yes | 40%/62x | LOF | - |
| CREBBP | NM_004380:exon4:c.T1101A:p.C367X Stopgain | Probably pathogenic | - | - | - | 50%/24x | LOF | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusakova, M.; Sharko, F.; Boulygina, E.; Slobodova, N.; Gladysheva-Azgari, M.; Badmazhapova, D.; Bullikh, A.; Khestanova, M.; Gabeeva, N.; Obukhova, T.; et al. High Tumor Mutation Burden (TMB) and a Novel Somatic Mutation in the TREX1 Gene in a Patient with Aggressive and Refractory High-Grade B-Cell Lymphoma: A Case Report. Int. J. Mol. Sci. 2025, 26, 2926. https://doi.org/10.3390/ijms26072926
Gusakova M, Sharko F, Boulygina E, Slobodova N, Gladysheva-Azgari M, Badmazhapova D, Bullikh A, Khestanova M, Gabeeva N, Obukhova T, et al. High Tumor Mutation Burden (TMB) and a Novel Somatic Mutation in the TREX1 Gene in a Patient with Aggressive and Refractory High-Grade B-Cell Lymphoma: A Case Report. International Journal of Molecular Sciences. 2025; 26(7):2926. https://doi.org/10.3390/ijms26072926
Chicago/Turabian StyleGusakova, Mariia, Fedor Sharko, Eugenia Boulygina, Natalia Slobodova, Maria Gladysheva-Azgari, Darima Badmazhapova, Artem Bullikh, Marina Khestanova, Nelli Gabeeva, Tatiana Obukhova, and et al. 2025. "High Tumor Mutation Burden (TMB) and a Novel Somatic Mutation in the TREX1 Gene in a Patient with Aggressive and Refractory High-Grade B-Cell Lymphoma: A Case Report" International Journal of Molecular Sciences 26, no. 7: 2926. https://doi.org/10.3390/ijms26072926
APA StyleGusakova, M., Sharko, F., Boulygina, E., Slobodova, N., Gladysheva-Azgari, M., Badmazhapova, D., Bullikh, A., Khestanova, M., Gabeeva, N., Obukhova, T., Zvonkov, E., & Tsygankova, S. (2025). High Tumor Mutation Burden (TMB) and a Novel Somatic Mutation in the TREX1 Gene in a Patient with Aggressive and Refractory High-Grade B-Cell Lymphoma: A Case Report. International Journal of Molecular Sciences, 26(7), 2926. https://doi.org/10.3390/ijms26072926