
Genomic Selection for Pea Grain Yield and Protein Content in Italian Environments for Target and Non-Target Genetic Bases
 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Data Analysis of the Genomic Selection Validation Set
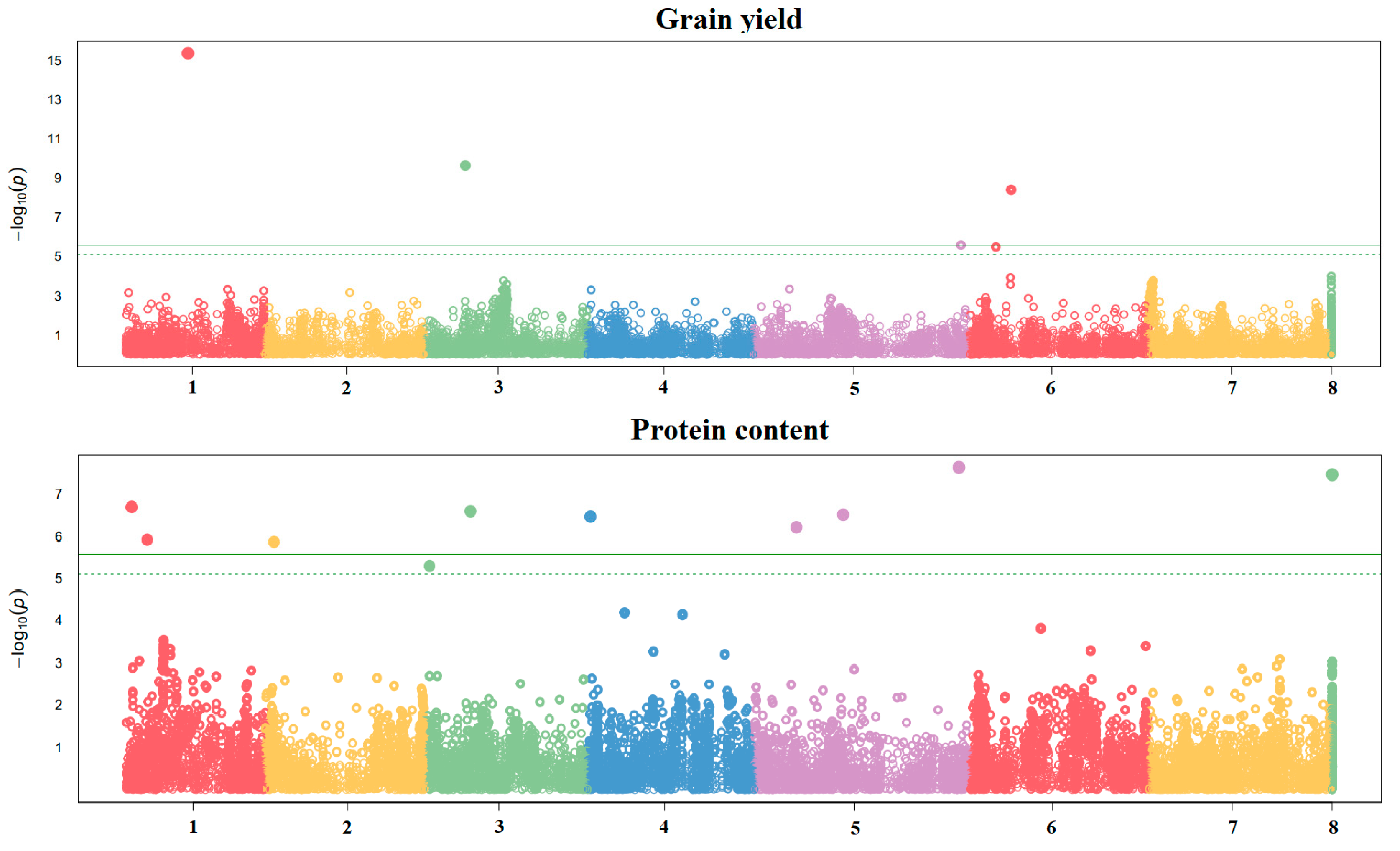
2.2. Genome-Wide Association Study
2.3. Genomic Selection
3. Discussion
4. Materials and Methods
4.1. Plant Material and Phenotyping
4.2. Phenotypic Data Analysis of the Genomic Selection Validation Set
4.3. Genotyping and Genomic Data Processing
4.4. Genome-Wide Association Study
4.5. Genomic Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GEI | Genotype × environment interaction |
| QTL | Quantitative trait locus |
| GS | Genomic selection |
| GBS | Genotyping by sequencing |
| RIL | Recombinant Inbred Line |
| SNP | Single-Nucleotide Polymorphism |
| NIRS | Near-infrared spectroscopy |
| MAF | Minor allele frequency |
References
- Pilorgé, E.; Muel, F. What vegetable oils and proteins for 2030? Would the protein fraction be the future of oil and protein crops? OCL 2016, 23, D402. [Google Scholar] [CrossRef]
- Nemecek, T.; Von Richthofen, J.S.; Dubois, G.; Casta, P.; Charles, R.; Pahl, H. Environmental impact of introducing grain legumes into European crop rotations. Eur. J. Agron. 2008, 28, 380–393. [Google Scholar] [CrossRef]
- Billen, G.; Lassaletta, L.; Garnier, J. A biogeochemical view of the global agro-food system: Nitrogen flows associated with protein production, consumption and trade. Glob. Food Secur. 2014, 3, 209–219. [Google Scholar] [CrossRef]
- Foyer, C.H.; Lam, H.-M.; Nguyen, H.T.; Siddique, K.H.M.; Varshney, R.K.; Colmer, T.D.; Cowling, W.; Bramley, H.; Mori, T.A.; Hodgson, J.M.; et al. Neglecting legumes has compromised human health and sustainable food production. Nat. Plants 2016, 2, 16112. [Google Scholar] [CrossRef]
- European Commission. Available online: https://agriculture.ec.europa.eu/document/download/fd08ade5-af88-4640-b9b0-b24ff2ca1e1e_en?filename=ext-eval-protein-crops-synth-sum_2009_en.pdf (accessed on 3 January 2025).
- De Visser, C.L.M.; Schreuder, R.; Stoddard, F. The EU’s dependency on soya bean import for the animal feed industry and potential for EU produced alternatives. Oilseeds Fats Crops Lipids 2014, 21, D407. [Google Scholar] [CrossRef]
- Carrouée, B.; Crépon, K.; Peyronnet, C. Les protéagineux: Intéret dans les systèmes de production fourragers francais et européens. Fourrages 2003, 174, 163–182. [Google Scholar]
- Annicchiarico, P. Adaptation of Cool-Season Grain Legume Species across Climatically-Contrasting Environments of Southern Europe. Agron. J. 2008, 100, 1647–1654. [Google Scholar] [CrossRef]
- Warkentin, T.; Smykal, P.; Coyne, C.J.; Weeden, N.; Domoney, C.; Bing, D.; Leonforte, A.; Xuxiao, Z.; Dixit, G.P.; Boros, L.; et al. Pea (Pisum sativum L.). In Handbook of Plant Breeding: Grain Legumes; De Ron, A., Ed.; Springer Science and Business Media: New York, NY, USA, 2015; pp. 37–83. [Google Scholar]
- Legume Hub. Available online: https://www.legumehub.eu/wp-content/uploads/2021/06/Legume-Futures-Report-1.2.pdf (accessed on 3 January 2025).
- Sagan, M.; Ney, B.; Duc, G. Plant symbiotic mutants as a tool to analyse nitrogen nutrition and yield relationship in field-growth peas (Pisum sativum L.). Plant Soil 1993, 153, 33–45. [Google Scholar] [CrossRef]
- Cousin, R. Peas (Pisum sativum L.). Field Crop. Res. 1997, 53, 111–130. [Google Scholar] [CrossRef]
- Annicchiarico, P.; Iannucci, A. Adaptation strategy, germplasm type and adaptive traits for field pea improvement in Italy based on variety responses across climatically contrasting environments. Field Crop. Res. 2008, 108, 133–142. [Google Scholar] [CrossRef]
- Iglesias-García, R.; Prats, E.; Flores, F.; Amri, M.; Mikić, A.; Rubiales, D. Assessment of field pea (Pisum sativum L.) grain yield, aerial biomass and flowering date stability in Mediterranean environments. Crop. Pasture Sci. 2017, 68, 915–923. [Google Scholar] [CrossRef]
- Bähr, M.; Fechner, A.; Hasenkopf, K.; Mittermaier, S.; Jahreis, G. Chemical composition of dehulled seeds of selected lupin cultivars in comparison to pea and soya bean. LWT 2014, 59, 587–590. [Google Scholar] [CrossRef]
- Burstin, J.; Gallardo, K.; Mir, R.R.; Varshney, R.K.; Duc, G. Improving protein content and nutrition quality. In Biology and Breeding of Food Legumes, 1st ed.; Pratap, A., Kumar, J., Eds.; CAB International: Wallingford, UK, 2011; pp. 314–328. [Google Scholar] [CrossRef]
- Duc, G.; Agrama, H.; Bao, S.; Berger, J.; Bourion, V.; De Ron, A.M.; Gowda, C.L.L.; Mikic, A.; Millot, D.; Singh, K.B.; et al. Breeding Annual Grain Legumes for Sustainable Agriculture: New Methods to Approach Complex Traits and Target New Cultivar Ideotypes. Crit. Rev. Plant Sci. 2014, 34, 381–411. [Google Scholar] [CrossRef]
- Crosta, M.; Nazzicari, N.; Ferrari, B.; Pecetti, L.; Russi, L.; Romani, M.; Cabassi, G.; Cavalli, D.; Marocco, A.; Annicchiarico, P. Pea Grain Protein Content Across Italian Environments: Genetic Relationship With Grain Yield, and Opportunities for Genome-Enabled Selection for Protein Yield. Front. Plant Sci. 2022, 12, 718713. [Google Scholar] [CrossRef] [PubMed]
- Cousin, R.; Messager, A.; Vingère, A. Breeding for yield in combining peas. In The Pea Crop; Hebblethwaite, P.D., Heath, M.C., Dawkins, T.C.K., Eds.; Butterworth-Heinemann: London, UK, 1985; pp. 115–129. [Google Scholar]
- Annicchiarico, P.; Nazzicari, N.; Pecetti, L.; Romani, M.; Ferrari, B.; Wei, Y.; Brummer, E.C. GBS-Based Genomic Selection for Pea Grain Yield under Severe Terminal Drought. Plant Genome 2017, 10, plantgenome2016.07.0072. [Google Scholar] [CrossRef]
- Bărbieru, A. Correlations between yield and several traits in a set of winter pea cultivars. Rom. Agric. Res. 2021, 38, 2021–2045. [Google Scholar]
- Crosta, M. Development and Validation of Genomic Selection Models for the Improvement of Pea Grain Yield and Protein Content in Italian Environments. Ph.D. Thesis, Catholic University of Sacred Hearth, Piacenza, Italy, 27 May 2024. [Google Scholar]
- Irzykowska, L.; Wolko, B. Interval mapping of QTLs controlling yield-related traits and grain protein content in Pisum sativum. J. Appl. Genet. 2004, 45, 297–306. [Google Scholar] [PubMed]
- Tar’An, B.; Warkentin, T.; Somers, D.; Miranda, D.; Vandenberg, A.; Blade, S.; Bing, D. Identification of quantitative trait loci for grain yield, seed protein concentration and maturity in field pea (Pisum sativum L.). Euphytica 2004, 136, 297–306. [Google Scholar] [CrossRef]
- Burstin, J.; Marget, P.; Huart, M.; Moessner, A.; Mangin, B.; Duchene, C.; Desprez, B.; Munier-Jolain, N.; Duc, G. Developmental Genes Have Pleiotropic Effects on Plant Morphology and Source Capacity, Eventually Impacting on Seed Protein Content and Productivity in Pea. Plant Physiol. 2007, 144, 768–781. [Google Scholar] [CrossRef]
- Krajewski, P.; Bocianowski, J.; Gawłowska, M.; Kaczmarek, Z.; Pniewski, T.; Święcicki, W.; Wolko, B. QTL for yield components and protein content: A multienvironment study of two pea (Pisum sativum L.) populations. Euphytica 2011, 183, 323–336. [Google Scholar] [CrossRef]
- Klein, A.; Houtin, H.; Rond, C.; Marget, P.; Jacquin, F.; Boucherot, K.; Huart, M.; Rivière, N.; Boutet, G.; Lejeune-Hénaut, I.; et al. QTL analysis of frost damage in pea suggests different mechanisms involved in frost tolerance. Theor. Appl. Genet. 2014, 127, 1319–1330. [Google Scholar] [CrossRef]
- Gali, K.K.; Liu, Y.; Sindhu, A.; Diapari, M.; Shunmugam, A.S.K.; Arganosa, G.; Daba, K.; Caron, C.; Lachagari, R.V.B.; Tar’an, B.; et al. Construction of high-density linkage maps for mapping quantitative trait loci for multiple traits in field pea (Pisum sativum L.). BMC Plant Biol. 2018, 18, 172. [Google Scholar] [CrossRef]
- Gali, K.K.; Sackville, A.; Tafesse, E.G.; Lachagari, V.R.; McPhee, K.; Hybl, M.; Mikić, A.; Smýkal, P.; McGee, R.; Burstin, J.; et al. Genome-Wide Association Mapping for Agronomic and Seed Quality Traits of Field Pea (Pisum sativum L.). Front. Plant Sci. 2019, 10, 1538. [Google Scholar] [CrossRef]
- Klein, A.; Houtin, H.; Rond-Coissieux, C.; Naudet-Huart, M.; Touratier, M.; Marget, P.; Burstin, J. Meta-analysis of QTL reveals the genetic control of yield-related traits and seed protein content in pea. Sci. Rep. 2020, 10, 15925. [Google Scholar] [CrossRef] [PubMed]
- Crosta, M.; Romani, M.; Nazzicari, N.; Ferrari, B.; Annicchiarico, P. Genomic prediction and allele mining of agronomic and morphological traits in pea (Pisum sativum) germplasm collections. Front. Plant Sci. 2023, 14, 1320506. [Google Scholar] [CrossRef]
- Bourion, V.; Rizvi, S.M.H.; Fournier, S.; de Larambergue, H.; Galmiche, F.; Marget, P.; Duc, G.; Burstin, J. Genetic dissection of nitrogen nutrition in pea through a QTL approach of root, nodule, and shoot variability. Theor. Appl. Genet. 2010, 121, 71–86. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Bernardo, R.; Yu, J. Prospects for genome-wide selection for quantitative traits in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; De Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.-A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Burstin, J.; Salloignon, P.; Chabert-Martinello, M.; Magnin-Robert, J.-B.; Siol, M.; Jacquin, F.; Chauveau, A.; Pont, C.; Aubert, G.; Delaitre, C.; et al. Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genom. 2015, 16, 105. [Google Scholar] [CrossRef]
- Tayeh, N.; Klein, A.; Le Paslier, M.-C.; Jacquin, F.; Houtin, H.; Rond, C.; Chabert-Martinello, M.; Magnin-Robert, J.-B.; Marget, P.; Aubert, G.; et al. Genomic Prediction in Pea: Effect of Marker Density and Training Population Size and Composition on Prediction Accuracy. Front. Plant Sci. 2015, 6, 941. [Google Scholar] [CrossRef] [PubMed]
- Al Bari, A.; Zheng, P.; Viera, I.; Worral, H.; Szwiec, S.; Ma, Y.; Main, D.; Coyne, C.J.; McGee, R.J.; Bandillo, N. Harnessing Genetic Diversity in the USDA Pea Germplasm Collection Through Genomic Prediction. Front. Genet. 2021, 12, 707754. [Google Scholar] [CrossRef]
- Annicchiarico, P.; Nazzicari, N.; Pecetti, L.; Romani, M.; Russi, L. Pea genomic selection for Italian environments. BMC Genom. 2019, 20, 603. [Google Scholar] [CrossRef] [PubMed]
- Annicchiarico, P.; Nazzicari, N.; Laouar, M.; Thami-Alami, I.; Romani, M.; Pecetti, L. Development and Proof-of-Concept Application of Genome-Enabled Selection for Pea Grain Yield under Severe Terminal Drought. Int. J. Mol. Sci. 2020, 21, 2414. [Google Scholar] [CrossRef]
- Saludares, R.A.; Atanda, S.A.; Piche, L.; Worral, H.; Dariva, F.; McPhee, K.; Bandillo, N. Multi-trait multi-environment genomic prediction of preliminary yield trial in pulse crop. Plant Genome 2024, 17, e20496. [Google Scholar] [CrossRef]
- Carpenter, M.A.; Goulden, D.S.; Woods, C.J.; Thomson, S.J.; Kenel, F.; Frew, T.J.; Cooper, R.D.; Timmerman-Vaughan, G.M. Genomic Selection for Ascochyta Blight Resistance in Pea. Front. Plant Sci. 2018, 9, 1878. [Google Scholar] [CrossRef]
- Osuna-Caballero, S.; Rubiales, D.; Annicchiarico, P.; Nazzicari, N.; Rispail, N. Genomic prediction for rust resistance in pea. Front. Plant Sci. 2024, 15, 1429802. [Google Scholar] [CrossRef]
- Atanda, S.A.; Steffes, J.; Lan, Y.; Al Bari, A.; Kim, J.; Morales, M.; Johnson, J.P.; Saludares, R.; Worral, H.; Piche, L.; et al. Multi-trait genomic prediction improves selection accuracy for enhancing seed mineral concentrations in pea. Plant Genome 2022, 15, e20260. [Google Scholar] [CrossRef]
- Duhnen, A.; Gras, A.; Teyssèdre, S.; Romestant, M.; Claustres, B.; Daydé, J.; Mangin, B. Genomic Selection for Yield and Seed Protein Content in Soybean: A Study of Breeding Program Data and Assessment of Prediction Accuracy. Crop Sci. 2017, 57, 1325–1337. [Google Scholar] [CrossRef]
- Stewart-Brown, B.B.; Song, Q.; Vaughn, J.N.; Li, Z. Genomic Selection for Yield and Seed Composition Traits Within an Applied Soybean Breeding Program. G3 Genes Genomes Genet. 2019, 9, 2253–2265. [Google Scholar] [CrossRef]
- Larkin, D.L.; Lozada, D.N.; Mason, R.E. Genomic Selection—Considerations for Successful Implementation in Wheat Breeding Programs. Agronomy 2019, 9, 479. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Rychel, S.; Nelson, M.N.; Wyrwa, K.; Naganowska, B.; Wolko, B. Expansion of the phosphatidylethanolamine binding protein family in legumes: A case study of Lupinus angustifolius L. FLOWERING LOCUS T homologs, LanFTc1 and LanFTc2. BMC Genom. 2016, 17, 820. [Google Scholar] [CrossRef] [PubMed]
- Dröge-Laser, W.; Snoek, B.L.; Snel, B.; Weiste, C. The Arabidopsis bZIP transcription factor family-an update. Curr. Opin. Plant Biol. 2018, 45, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Qiao, Z.; Li, Y.; Yang, Z.; Wang, C.; Zhang, Y.; Liu, L.; Wang, B. RING zinc finger proteins in plant abiotic stress tolerance. Front. Plant Sci. 2022, 13, 877011. [Google Scholar] [CrossRef]
- Brito, D.S.; Quinhones, C.G.; Neri-Silva, R.; Heinemann, B.; Schertl, P.; Cavalcanti, J.H.F.; Eubel, H.; Hildebrandt, T.; Nunes-Nesi, A.; Braun, H.-P.; et al. The role of the electron-transfer flavoprotein: Ubiquinone oxidoreductase following carbohydrate starvation in Arabidopsis cell cultures. Plant Cell Rep. 2022, 41, 431–446. [Google Scholar] [CrossRef]
- Gust, A.A.; Willmann, R.; Desaki, Y.; Grabherr, H.M.; Nürnberger, T. Plant LysM proteins: Modules mediating symbiosis and immunity. Trends Plant Sci. 2012, 17, 495–502. [Google Scholar] [CrossRef]
- Sultana, N.; Islam, S.; Juhasz, A.; Ma, W. Wheat leaf senescence and its regulatory gene network. Crop J. 2021, 9, 703–717. [Google Scholar] [CrossRef]
- Annicchiarico, P. Scelta varietale in pisello e favino rispetto all’ambiente e all’utilizzo. Inf. Agrar. 2005, 61, 47–52. [Google Scholar]
- Annicchiarico, P.; Filippi, L. A Field Pea Ideotype for Organic Systems of Northern Italy. J. Crop Improv. 2007, 20, 193–203. [Google Scholar] [CrossRef]
- Annicchiarico, P.; Nazzicari, N.; Notario, T.; Martin, C.M.; Romani, M.; Ferrari, B.; Pecetti, L. Pea Breeding for Intercropping With Cereals: Variation for Competitive Ability and Associated Traits, and Assessment of Phenotypic and Genomic Selection Strategies. Front. Plant Sci. 2021, 12, 731949. [Google Scholar] [CrossRef] [PubMed]
- Puritz, J.B.; Hollenbeck, C.M.; Gold, J.R. dDocent: A RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2014, 2, e431. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Stekhoven, D.J.; Bühlmann, P. MissForest—Non-parametric missing value imputation for mixed-type data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef]
- Andridge, R.R.; Little, R.J. A review of hot deck imputation for survey non-response. Int. Stat. Rev. 2010, 78, 40–64. [Google Scholar] [CrossRef]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome wide association studies with both individuals and markers in the millions. GigaScience 2019, 8, 154. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Wang, M.; Xu, S. Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity 2019, 123, 287–306. [Google Scholar] [CrossRef]
- Yendle, P.W.; MacFie, H.J. Discriminant principal components analysis. J. Chemom. 1989, 3, 589–600. [Google Scholar] [CrossRef]
- Laurie, C.C.; Doheny, K.F.; Mirel, D.B.; Pugh, E.W.; Bierut, L.J.; Bhangale, T.; Boehm, F.; Caporaso, N.E.; Cornelis, M.C.; Edenberg, H.J.; et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol. 2010, 34, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pazaran, G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]
- Marroni, F.; Pinosio, S.; Zaina, G.; Fogolari, F.; Felice, N.; Cattonaro, F.; Morgante, M. Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genet. Genomes 2011, 7, 1011–1023. [Google Scholar] [CrossRef]

{kind=link}
| RIL Population | |||||||
|---|---|---|---|---|---|---|---|
| Trait | Statistic | A × I | K × I | D × A | A × G | C × I | Mean |
| GY | Mean a | 6.20 a | 6.18 a | 5.46 b | 5.36 b | 5.12 b | 5.66 |
| GY | CV1 | 12.9 | 27.9 | 25.9 | 24.5 | 32.8 | 24.8 |
| GY | CV2 | 16.4 | 17.5 | 18.6 | 26.9 | 23.2 | 20.5 |
| GY | r b | 0.41 + | 0.57 ** | 0.42 + | 0.42 * | 0.08 NS | 0.38 |
| PC | Mean a | 22.74 c | 23.28 b | 22.53 c | 23.80 a | 22.54 c | 23.0 |
| PC | CV1 | 4.2 | 6.0 | 6.1 | 7.0 | 6.5 | 6.0 |
| PC | CV2 | 4.4 | 4.3 | 4.7 | 3.7 | 27.3 | 8.9 |
| PC | r b | 0.49 * | 0.65 *** | 0.58 ** | 0.44 * | 0.43 + | 0.52 |
| PY | Mean a | 1.42 a | 1.46 a | 1.24 bc | 1.28 b | 1.17 c | 1.31 |
| PY | CV1 | 13.6 | 30.9 | 26.9 | 24.9 | 34.1 | 26.1 |
| PY | CV2 | 17.8 | 19.5 | 19.8 | 28.1 | 37.2 | 24.5 |
| PY | r b | 0.50 * | 0.60 ** | 0.49 * | 0.52 * | 0.21 NS | 0.46 |
| Predictive Ability | ||||||
|---|---|---|---|---|---|---|
| Within RILs | Across RILs | |||||
| Trait | Year | Target GB | Non-Target GB | Target GB | Non-Target GB | |
| Grain yield | 2018–2019 | 0.439 | 0.113 | 0.359 | −0.087 | |
| Grain yield | 2019–2020 | 0.458 | 0.011 | 0.525 | −0.100 | |
| Grain yield | mean | 0.505 | 0.079 | 0.480 | −0.110 | |
| Protein content | 2018–2019 | 0.534 | 0.372 | 0.560 | 0.295 | |
| Protein content | 2019–2020 | 0.675 | 0.314 | 0.632 | 0.117 | |
| Protein content | mean | 0.673 | 0.360 | 0.663 | 0.229 | |
| Protein yield | 2018–2019 | 0.452 | 0.085 | 0.400 | −0.155 | |
| Protein yield | 2019–2020 | 0.490 | −0.089 | 0.572 | −0.269 | |
| Protein yield | mean | 0.514 | 0.003 | 0.513 | −0.256 | |
| Predictive Ability | |||||||
|---|---|---|---|---|---|---|---|
| Target GB | Non-Target GB | ||||||
| Trait | Year | A × I | K × I | D × A | A × G | C × I | |
| Grain yield | 2018–2019 | 0.368 | 0.510 | −0.063 | 0.147 | 0.256 | |
| Grain yield | 2019–2020 | 0.303 | 0.613 | −0.149 | 0.047 | 0.136 | |
| Grain yield | mean | 0.407 | 0.603 | −0.100 | 0.104 | 0.233 | |
| Protein content | 2018–2019 | 0.575 | 0.492 | 0.202 | 0.721 | 0.195 | |
| Protein content | 2019–2020 | 0.636 | 0.714 | 0.322 | 0.288 | 0.331 | |
| Protein content | mean | 0.708 | 0.639 | 0.385 | 0.663 | 0.030 | |
| Protein yield | 2018–2019 | 0.387 | 0.518 | −0.094 | 0.111 | 0.237 | |
| Protein yield | 2019–2020 | 0.319 | 0.662 | −0.186 | 0.030 | −0.111 | |
| Protein yield | mean | 0.412 | 0.616 | −0.167 | 0.074 | 0.101 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crosta, M.; Nazzicari, N.; Pecetti, L.; Notario, T.; Romani, M.; Ferrari, B.; Cabassi, G.; Annicchiarico, P. Genomic Selection for Pea Grain Yield and Protein Content in Italian Environments for Target and Non-Target Genetic Bases. Int. J. Mol. Sci. 2025, 26, 2991. https://doi.org/10.3390/ijms26072991
Crosta M, Nazzicari N, Pecetti L, Notario T, Romani M, Ferrari B, Cabassi G, Annicchiarico P. Genomic Selection for Pea Grain Yield and Protein Content in Italian Environments for Target and Non-Target Genetic Bases. International Journal of Molecular Sciences. 2025; 26(7):2991. https://doi.org/10.3390/ijms26072991
Chicago/Turabian StyleCrosta, Margherita, Nelson Nazzicari, Luciano Pecetti, Tommaso Notario, Massimo Romani, Barbara Ferrari, Giovanni Cabassi, and Paolo Annicchiarico. 2025. "Genomic Selection for Pea Grain Yield and Protein Content in Italian Environments for Target and Non-Target Genetic Bases" International Journal of Molecular Sciences 26, no. 7: 2991. https://doi.org/10.3390/ijms26072991
APA StyleCrosta, M., Nazzicari, N., Pecetti, L., Notario, T., Romani, M., Ferrari, B., Cabassi, G., & Annicchiarico, P. (2025). Genomic Selection for Pea Grain Yield and Protein Content in Italian Environments for Target and Non-Target Genetic Bases. International Journal of Molecular Sciences, 26(7), 2991. https://doi.org/10.3390/ijms26072991