

Association of Complement Proteins with C Reactive Protein in Non-Obese Women with and Without Polycystic Ovary Syndrome
 , , ,
, , ,  , and
, and
Abstract
1. Introduction
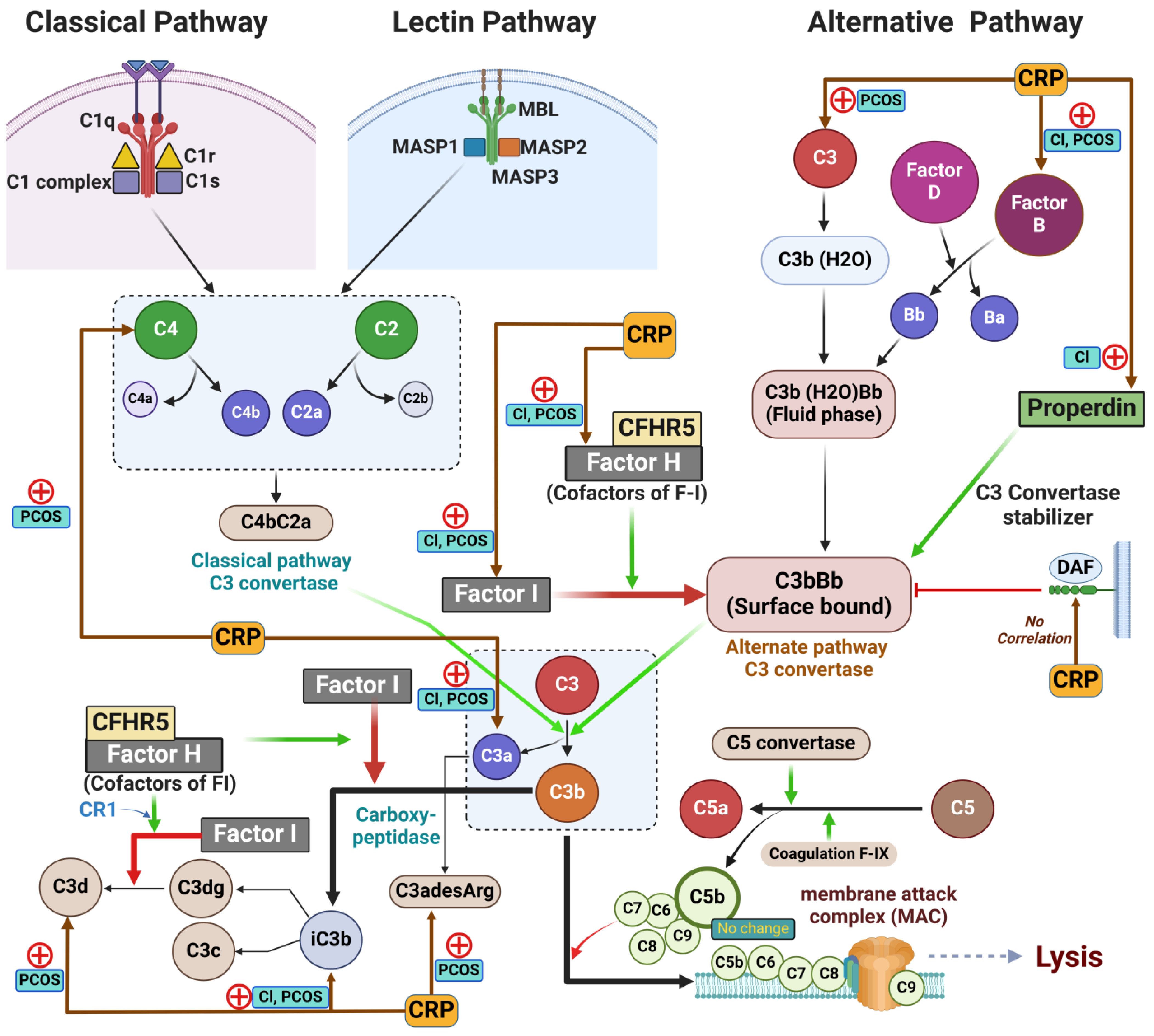
2. Results
2.1. Demographics and Biochemical Measurements
2.2. Complement Protein Activation in Non-Obese Women with and Without PCOS [9,34]
2.3. Correlations of CRP and Hyperandrogenemia with BMI
2.4. Correlations Between CRP and Complement Proteins in Non-Obese Women with and Without PCOS
2.5. Correlations of Complement Activation Related Proteins with Hyperandrogenemia, and BMI [34]
3. Discussion
4. Materials and Methods
4.1. Sample Analysis
4.2. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCOS | Polycystic ovary syndrome |
| CRP | C-reactive protein |
| BMI | Body mass index |
| MBL | Mannose-binding lectin |
| MAC | Membrane attack complex |
| SOMA | Slow Off-rate Modified Aptamer |
| HOMA-IR | Homeostasis model assessment of insulin resistance |
| SHBG | Sex hormone binding globulin |
| FAI | Free androgen index |
| AMH | Anti-Müllerian hormone |
| DAF | Decay-accelerating factor |
| CFHR5 | Complement factor H-related protein 5 |
References
- Sathyapalan, T.; Atkin, S.L. Recent advances in cardiovascular aspects of polycystic ovary syndrome. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2012, 166, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Complement activation: An emerging player in the pathogenesis of cardiovascular disease. Scientifica 2012, 2012, 402783. [Google Scholar] [CrossRef]
- Sathyapalan, T.; Shepherd, J.; Coady, A.M.; Kilpatrick, E.S.; Atkin, S.L. Atorvastatin reduces malondialdehyde concentrations in patients with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 3951–3955. [Google Scholar] [PubMed]
- Bannigida, D.M.; Nayak, B.S.; Vijayaraghavan, R. Insulin resistance and oxidative marker in women with PCOS. Arch. Physiol. Biochem. 2020, 126, 183–186. [Google Scholar]
- Niinuma, S.A.; Lubbad, L.; Lubbad, W.; Moin, A.S.M.; Butler, A.E. The Role of Heat Shock Proteins in the Pathogenesis of Polycystic Ovarian Syndrome: A Review of the Literature. Int. J. Mol. Sci. 2023, 24, 1838. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.S.M.; Sathyapalan, T.; Butler, A.E.; Atkin, S.L. Coagulation factor dysregulation in polycystic ovary syndrome is an epiphenomenon of obesity. Clin. Endocrinol. 2023, 98, 796–802. [Google Scholar]
- Lewis, R.D.; Narayanaswamy, A.K.; Farewell, D.; Rees, D.A. Complement activation in polycystic ovary syndrome occurs in the postprandial and fasted state and is influenced by obesity and insulin sensitivity. Clin. Endocrinol. 2021, 94, 74–84. [Google Scholar]
- Yang, S.; Li, Q.; Song, Y.; Tian, B.; Cheng, Q.; Qing, H.; Zhong, L.; Xia, W. Serum complement C3 has a stronger association with insulin resistance than high-sensitivity C-reactive protein in women with polycystic ovary syndrome. Fertil. Steril. 2011, 95, 1749–1753. [Google Scholar]
- Moin, A.S.M.; Sathyapalan, T.; Butler, A.E.; Atkin, S.L. Classical and alternate complement factor overexpression in non-obese weight matched women with polycystic ovary syndrome does not correlate with vitamin D. Front. Endocrinol. 2022, 13, 935750. [Google Scholar]
- Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. Components of the Complement Cascade Differ in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2022, 23, 12232. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar]
- Janeway, C.A., Jr.; Ttravers, P.; Walport, M.; Shlomchik, M. Immunobiology: The Immune System in Health and Disease, 5th ed.Garland Publishing: New York, NY, USA, 2001. [Google Scholar]
- Shim, K.; Begum, R.; Yang, C.; Wang, H. Complement activation in obesity, insulin resistance, and type 2 diabetes mellitus. World J. Diabetes 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, G.; Hedblad, B.; Eriksson, K.-F.; Janzon, L.; Lindgärde, F. Complement C3 Is a Risk Factor for the Development of Diabetes: A Population-Based Cohort Study. Diabetes 2005, 54, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, M.; Hansen, T.; Flyvbjerg, A. Complement activation and cardiovascular disease. Horm. Metab. Res. 2008, 40, 626–634. [Google Scholar] [PubMed]
- Feng, D.; Shi, B.; Bi, F.; Sagnelli, M.; Sun, X.; Jiao, J.; Wang, X.; Li, D. Elevated Serum Mannose Levels as a Marker of Polycystic Ovary Syndrome. Front. Endocrinol. 2019, 10, 711. [Google Scholar]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar]
- Kushner, I.; Jiang, S.L.; Zhang, D.; Lozanski, G.; Samols, D. Do post-transcriptional mechanisms participate in induction of C-reactive protein and serum amyloid A by IL-6 and IL-1? Ann. N. Y. Acad. Sci. 1995, 762, 102–107. [Google Scholar]
- Zhou, H.H.; Tang, Y.L.; Xu, T.H.; Cheng, B. C-reactive protein: Structure, function, regulation, and role in clinical diseases. Front. Immunol. 2024, 15, 1425168. [Google Scholar]
- Banait, T.; Wanjari, A.; Danade, V.; Banait, S.; Jain, J. Role of High-Sensitivity C-reactive Protein (Hs-CRP) in Non-communicable Diseases: A Review. Cureus 2022, 14, e30225. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar]
- Kaplan, M.H.; Volanakis, J.E. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J. Immunol. 1974, 112, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.; Rent, R.; Gewurz, H. Interactions of C-reactive protein with the complement system. I. Protamine-induced consumption of complement in acute phase sera. J. Exp. Med. 1974, 140, 631–647. [Google Scholar] [PubMed]
- Noone, D.P.; Isendoorn, M.M.E.; Hamers, S.M.W.R.; Keizer, M.E.; Wulffelé, J.; van der Velden, T.T.; Dijkstra, D.J.; Trouw, L.A.; Filippov, D.V.; Sharp, T.H. Structural basis for surface activation of the classical complement cascade by the short pentraxin C-reactive protein. Proc. Natl. Acad. Sci. USA 2024, 121, e2404542121. [Google Scholar]
- Mold, C.; Gewurz, H. Inhibitory effect of C-reactive protein on alternative C pathway activation by liposomes and Streptococcus pneumoniae. J. Immunol. 1981, 127, 2089–2092. [Google Scholar]
- Mold, C.; Kingzette, M.; Gewurz, H. C-reactive protein inhibits pneumococcal activation of the alternative pathway by increasing the interaction between factor H and C3b. J. Immunol. 1984, 133, 882–885. [Google Scholar]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar]
- Rizo-Téllez, S.A.; Sekheri, M.; Filep, J.G. C-reactive protein: A target for therapy to reduce inflammation. Front. Immunol. 2023, 14, 1237729. [Google Scholar]
- Velija-Asimi, Z. C-reactive protein in obese PCOS women and the effect of metformin therapy. Bosn. J. Basic Med. Sci. 2007, 7, 90–93. [Google Scholar]
- Escobar-Morreale, H.F.; Luque-Ramírez, M.; González, F. Circulating inflammatory markers in polycystic ovary syndrome: A systematic review and metaanalysis. Fertil. Steril. 2011, 95, 1048–1058.e1-e2. [Google Scholar]
- Aboeldalyl, S.; James, C.; Seyam, E.; Ibrahim, E.M.; Shawki, H.E.; Amer, S. The Role of Chronic Inflammation in Polycystic Ovarian Syndrome-A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2021, 22, 2734. [Google Scholar] [CrossRef] [PubMed]
- Timpson, N.J.; Nordestgaard, B.G.; Harbord, R.M.; Zacho, J.; Frayling, T.M.; Tybjærg-Hansen, A.; Smith, G.D. C-reactive protein levels and body mass index: Elucidating direction of causation through reciprocal Mendelian randomization. Int. J. Obes. 2011, 35, 300–308. [Google Scholar]
- Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. Complement Dysregulation in Obese Versus Nonobese Polycystic Ovary Syndrome Patients. Cells 2023, 12, 2002. [Google Scholar] [CrossRef]
- Abdul-Ameer, F.; AlAsadi, I.J.A.; Hosseini, A.; Bahreini, E. The relationship between serum CTRP-5, C3a/desArg, and complement-C3 levels and hypothyroidism in women with polycystic ovary syndrome. BMC Endocr. Disord. 2024, 24, 272. [Google Scholar]
- Oktenli, C.; Ozgurtas, T.; Dede, M.; Sanisoglu, Y.S.; Yenen, M.C.; Yesilova, Z.; Kenar, L.; Kurt, Y.G.; Baser, I.; Smith, J.; et al. Metformin decreases circulating acylation-stimulating protein levels in polycystic ovary syndrome. Gynecol. Endocrinol. 2007, 23, 710–715. [Google Scholar] [PubMed]
- Wu, Y.; Zhang, J.; Wen, Y.; Wang, H.; Zhang, M.; Cianflone, K. Increased acylation-stimulating protein, C-reactive protein, and lipid levels in young women with polycystic ovary syndrome. Fertil. Steril. 2009, 91, 213–219. [Google Scholar]
- Dehdashtihaghighat, S.; Mehdizadehkashi, A.; Arbabi, A.; Pishgahroudsari, M.; Chaichian, S. Assessment of C-reactive Protein and C3 as Inflammatory Markers of Insulin Resistance in Women with Polycystic Ovary Syndrome: A Case-Control Study. J. Reprod. Infertil. 2013, 14, 197–201. [Google Scholar]
- Snyder, M.L.; Shields, K.J.; Korytkowski, M.T.; Sutton-Tyrrell, K.; Talbott, E.O. Complement protein C3 and coronary artery calcium in middle-aged women with polycystic ovary syndrome and controls. Gynecol. Endocrinol. 2014, 30, 511–515. [Google Scholar]
- Ramanjaneya, M.; Abdalhakam, I.; Bettahi, I.; Bensila, M.; Jerobin, J.; Aye, M.M.; Alkasem, M.; Sathyapalan, T.; Atkin, S.L.; Abou-Samra, A.-B. Effect of Moderate Aerobic Exercise on Complement Activation Pathways in Polycystic Ovary Syndrome Women. Front. Endocrinol. 2021, 12, 740703. [Google Scholar]
- Gursoy Calan, O.; Calan, M.; Yesil Senses, P.; Unal Kocabas, G.; Ozden, E.; Sari, K.R.; Kocar, M.; Imamoglu, C.; Senses, Y.M.; Bozkaya, G.; et al. Increased adipsin is associated with carotid intima media thickness and metabolic disturbances in polycystic ovary syndrome. Clin. Endocrinol. 2016, 85, 910–917. [Google Scholar]
- Wolbink, G.J.; Brouwer, M.C.; Buysmann, S.; ten Berge, I.J.; Hack, C.E. CRP-mediated activation of complement in vivo: Assessment by measuring circulating complement-C-reactive protein complexes. J. Immunol. 1996, 157, 473–479. [Google Scholar] [PubMed]
- Basile, U.; Bruno, C.; Napodano, C.; Vergani, E.; Gulli, F.; Piunno, G.; Pocino, K.; Stefanile, A.; Mancini, A. Evaluation of immunoglobulins subclasses and free-light chains in non-obese patients with polycystic ovary syndrome and correlations with hormonal and metabolic parameters: Preliminary data. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4198–4204. [Google Scholar] [PubMed]
- Nilsson, B.; Hamad, O.A.; Ahlström, H.; Kullberg, J.; Johansson, L.; Lindhagen, L.; Haenni, A.; Ekdahl, K.N.; Lind, L. C3 and C4 are strongly related to adipose tissue variables and cardiovascular risk factors. Eur. J. Clin. Investig. 2014, 44, 587–596. [Google Scholar]
- Cianflone, K.; Xia, Z.; Chen, L.Y. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim. Biophys. Acta (BBA) Biomembr. 2003, 1609, 127–143. [Google Scholar]
- Guo, Z.; Fan, X.; Yao, J.; Tomlinson, S.; Yuan, G.; He, S. The role of complement in nonalcoholic fatty liver disease. Front. Immunol. 2022, 13, 1017467. [Google Scholar]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement component C3—The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar]
- Pangburn, M.K.; Müller-Eberhard, H.J. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem. J. 1986, 235, 723–730. [Google Scholar]
- Farries, T.C.; Lachmann, P.J.; Harrison, R.A. Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem. J. 1988, 252, 47–54. [Google Scholar]
- Fearon, D.T.; Austen, K.F. Properdin: Binding to C3b and stabilization of the C3b-dependent C3 convertase. J. Exp. Med. 1975, 142, 856–863. [Google Scholar]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar]
- Hammond, D.J., Jr.; Singh, S.K.; Thompson, J.A.; Beeler, B.W.; Rusiñol, A.E.; Pangburn, M.K.; Potempa, L.A.; Agrawal, A. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J. Biol. Chem. 2010, 285, 36235–36244. [Google Scholar] [CrossRef] [PubMed]
- O’Flynn, J.; van der Pol, P.; Dixon, K.O.; Prohászka, Z.; Daha, M.R.; van Kooten, C. Monomeric C-reactive protein inhibits renal cell-directed complement activation mediated by properdin. Am. J. Physiol. Renal Physiol. 2016, 310, F1308–F1316. [Google Scholar] [CrossRef]
- Barnum, S.R.; Niemann, M.A.; Kearney, J.F.; Volanakis, J.E. Quantitation of complement factor D in human serum by a solid-phase radioimmunoassay. J. Immunol. Methods 1984, 67, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Cunnion, K.M.; Hair, P.S.; Buescher, E.S. Cleavage of complement C3b to iC3b on the surface of Staphylococcus aureus is mediated by serum complement factor I. Infect. Immun. 2004, 72, 2858–2863. [Google Scholar] [CrossRef] [PubMed]
- Vik, D.P.; MuñOz-Cánoves, P.; Chaplin, D.D.; Tack, B.F. The Third Component of Complement; Factor, H., Ed.; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Kajander, T.; Lehtinen, M.J.; Hyvärinen, S.; Bhattacharjee, A.; Leung, E.; Isenman, D.E.; Meri, S.; Goldman, A.; Jokiranta, T.S. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. USA 2011, 108, 2897–2902. [Google Scholar] [CrossRef]
- Dempsey, P.W.; Allison, M.E.; Akkaraju, S.; Goodnow, C.C.; Fearon, D.T. C3d of complement as a molecular adjuvant: Bridging innate and acquired immunity. Science 1996, 271, 348–350. [Google Scholar] [CrossRef]
- Skerka, C.; Chen, Q.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement factor H related proteins (CFHRs). Mol. Immunol. 2013, 56, 170–180. [Google Scholar] [CrossRef]
- McRae, J.L.; Duthy, T.G.; Griggs, K.M.; Ormsby, R.J.; Cowan, P.J.; Cromer, B.A.; McKinstry, W.J.; Parker, M.W.; Murphy, B.F.; Gordon, D.L. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J. Immunol. 2005, 174, 6250–6256. [Google Scholar] [CrossRef]
- Lu, X.; Li, Y.; Simovic, M.O.; Peckham, R.; Wang, Y.; Tsokos, G.C.; Lucca, J.J.D. Decay-accelerating factor attenuates C-reactive protein-potentiated tissue injury after mesenteric ischemia/reperfusion. J. Surg. Res. 2011, 167, e103–e115. [Google Scholar] [CrossRef]
- Kew, R.R.; Hyers, T.M.; Webster, R.O. Human C-reactive protein inhibits neutrophil chemotaxis in vitro: Possible implications for the adult respiratory distress syndrome. J. Lab. Clin. Med. 1990, 115, 339–345. [Google Scholar]
- Huber-Lang, M.; Younkin, E.M.; Sarma, J.V.; Riedemann, N.; McGuire, S.R.; Lu, K.T.; Kunkel, R.; Younger, J.G.; Zetoune, F.S.; Ward, P.A. Generation of C5a by phagocytic cells. Am. J. Pathol. 2002, 161, 1849–1859. [Google Scholar] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [PubMed]
- Engmann, L.; Jin, S.; Sun, F.; Legro, R.S.; Polotsky, A.J.; Hansen, K.R.; Coutifaris, C.; Diamond, M.P.; Eisenberg, E.; Zhang, H.; et al. Racial and ethnic differences in the polycystic ovary syndrome metabolic phenotype. Am. J. Obstet. Gynecol. 2017, 216, 493.e1–493.e13. [Google Scholar] [PubMed]
- Cunningham, T.K.; Allgar, V.; Dargham, S.R.; Kilpatrick, E.; Sathyapalan, T.; Maguiness, S.; Rudin, H.R.M.; Ghani, N.M.A.; Latiff, A.; Atkin, S.L. Association of Vitamin D Metabolites With Embryo Development and Fertilization in Women With and Without PCOS Undergoing Subfertility Treatment. Front. Endocrinol. 2019, 10, 13. [Google Scholar]
- Sathyapalan, T.; Al-Qaissi, A.; Kilpatrick, E.S.; Dargham, S.R.; Atkin, S.L. Anti-Mullerian hormone measurement for the diagnosis of polycystic ovary syndrome. Clin. Endocrinol. 2017, 88, 258–262. [Google Scholar]
- Sathyapalan, T.; Al-Qaissi, A.; Kilpatrick, E.S.; Dargham, S.R.; Adaway, J.; Keevil, B.; Atkin, S.L. Salivary testosterone measurement in women with and without polycystic ovary syndrome. Sci. Rep. 2017, 7, 3589. [Google Scholar] [CrossRef]
- Kahal, H.; Halama, A.; Aburima, A.; Bhagwat, A.M.; Butler, A.E.; Grauman, J.; Suhre, K.; Sathyapalan, T.; Atkin, S.L. Effect of induced hypoglycemia on inflammation and oxidative stress in type 2 diabetes and control subjects. Sci. Rep. 2020, 10, 4750. [Google Scholar] [CrossRef]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| PCOS (n = 24) | Control (n = 24) | |
|---|---|---|
| Age (years) | 31 ± 6.4 | 32.5 ± 4.1 |
| BMI (kg/m2) | 25.9 ± 1.8 | 24.8 ± 1.1 |
| Insulin (IU/mL) | 8.1 ± 4.7 | 7.7 ± 4.0 |
| HOMA-IR | 1.9 ± 1.6 | 1.8 ± 1.0 |
| Testosterone (nmol/L) | 1.4 ± 0.8 | 0.7 ± 0.4 *** |
| SHBG (nmol/L) | 71.7 ± 62.2 | 104 ± 80 |
| FAI | 4.1 ± 2.9 | 1.3 ± 0.5 ** |
| CRP (mg L−1) | 2.8 ± 2.6 | 2.3 ± 2.34 |
| AMH (ng/mL) | 57 ± 14 | 24 ± 13 ** |
| PCOS | Control | p Value | |||
|---|---|---|---|---|---|
| Alternative pathway | Proteins | C3 | 65,878 (26,872) | 45,742 (18,189) | 0.002 |
| C3a | 534 (204) | 415 (101) | 0.007 | ||
| C3adesArg | 152,050 (32,483) | 121,110 (45,753) | 0.004 | ||
| C3b | 50,982 (28,296) | 46,250 (39,450) | 0.6 | ||
| iC3b | 7148 (2127) | 5991 (1425) | 0.02 | ||
| C3d | 10,427 (3675) | 8207 (3261) | 0.02 | ||
| Negative regulators | Factor I | 44,861 (6786) | 39,960 (7356) | 0.01 | |
| Factor H | 60,898 (9191) | 59,289 (6016) | 0.43 | ||
| CFHR5 | 1454 (399) | 1677 (1416) | 0.42 | ||
| DAF (CD55) | 15,182 (3220) | 13,886 (3339) | 0.14 | ||
| Positive regulators | Factor B | 30,257 (6541) | 28,172 (5791) | 0.20 | |
| Factor D | 733 (99) | 693 (150) | 0.24 | ||
| Properdin | 152,592 (42,743) | 117,488 (50,041) | 0.006 | ||
| Classical pathway | Proteins | C1q | 35,146 (9269) | 35,294 (7886) | 0.95 |
| C1r | 3203 (886) | 3740 (3992) | 0.48 | ||
| C4 | 119,057 (28,429) | 104,245 (28,069) | 0.05 | ||
| C4a | 71,549 (9802) | 73,037 (2258) | 0.43 | ||
| C4b | 349 (186) | 335 (202) | 0.78 | ||
| Lectin pathway | Proteins | C2 | 2878 (319) | 2874 (228) | 0.96 |
| MBL | 12,058 (5835) | 13,322 (7385) | 0.47 | ||
| Terminal pathway | Proteins | C5 | 7267 (1007) | 6881 (1063) | 0.16 |
| C5a | 14,729 (5811) | 11,343 (4953) | 0.02 | ||
| C5b, 6 Complex | 521 (62) | 502 (62) | 0.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, A.E.; Moin, A.S.M.; Begam, H.H.; Waris, S.; Azeez, J.M.; Sathyapalan, T.; Atkin, S.L.; Brennan, E. Association of Complement Proteins with C Reactive Protein in Non-Obese Women with and Without Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2025, 26, 3008. https://doi.org/10.3390/ijms26073008
Butler AE, Moin ASM, Begam HH, Waris S, Azeez JM, Sathyapalan T, Atkin SL, Brennan E. Association of Complement Proteins with C Reactive Protein in Non-Obese Women with and Without Polycystic Ovary Syndrome. International Journal of Molecular Sciences. 2025; 26(7):3008. https://doi.org/10.3390/ijms26073008
Chicago/Turabian StyleButler, Alexandra E., Abu Saleh Md Moin, Hamna H. Begam, Sana Waris, Juberiya M. Azeez, Thozhukat Sathyapalan, Stephen L. Atkin, and Edwina Brennan. 2025. "Association of Complement Proteins with C Reactive Protein in Non-Obese Women with and Without Polycystic Ovary Syndrome" International Journal of Molecular Sciences 26, no. 7: 3008. https://doi.org/10.3390/ijms26073008
APA StyleButler, A. E., Moin, A. S. M., Begam, H. H., Waris, S., Azeez, J. M., Sathyapalan, T., Atkin, S. L., & Brennan, E. (2025). Association of Complement Proteins with C Reactive Protein in Non-Obese Women with and Without Polycystic Ovary Syndrome. International Journal of Molecular Sciences, 26(7), 3008. https://doi.org/10.3390/ijms26073008