
Convergence in Amino Acid Outsourcing Between Animals and Predatory Bacteria


Abstract
1. Introduction
2. Results
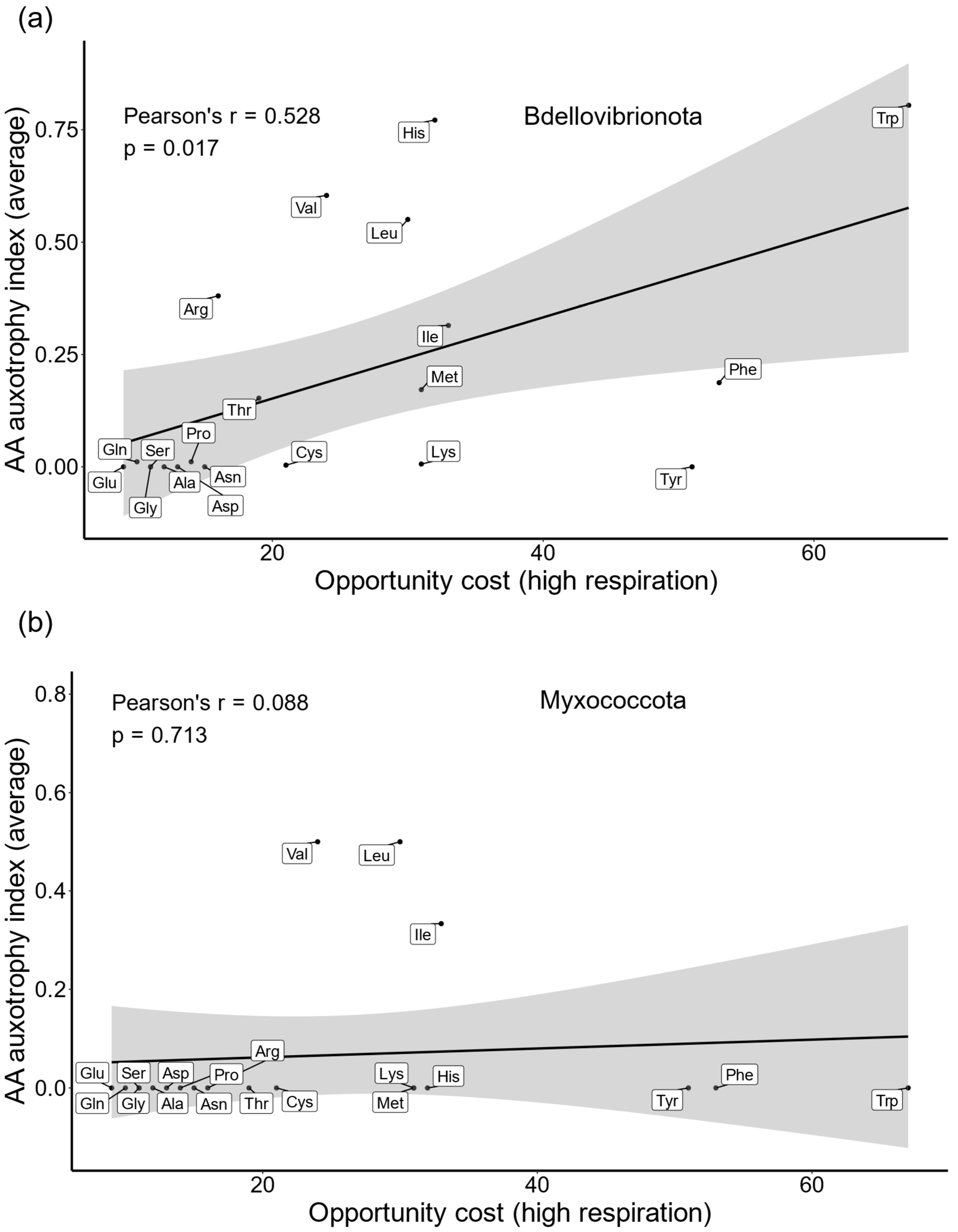
2.1. AA Auxotrophies in Bdellovibrionota and Myxococcota
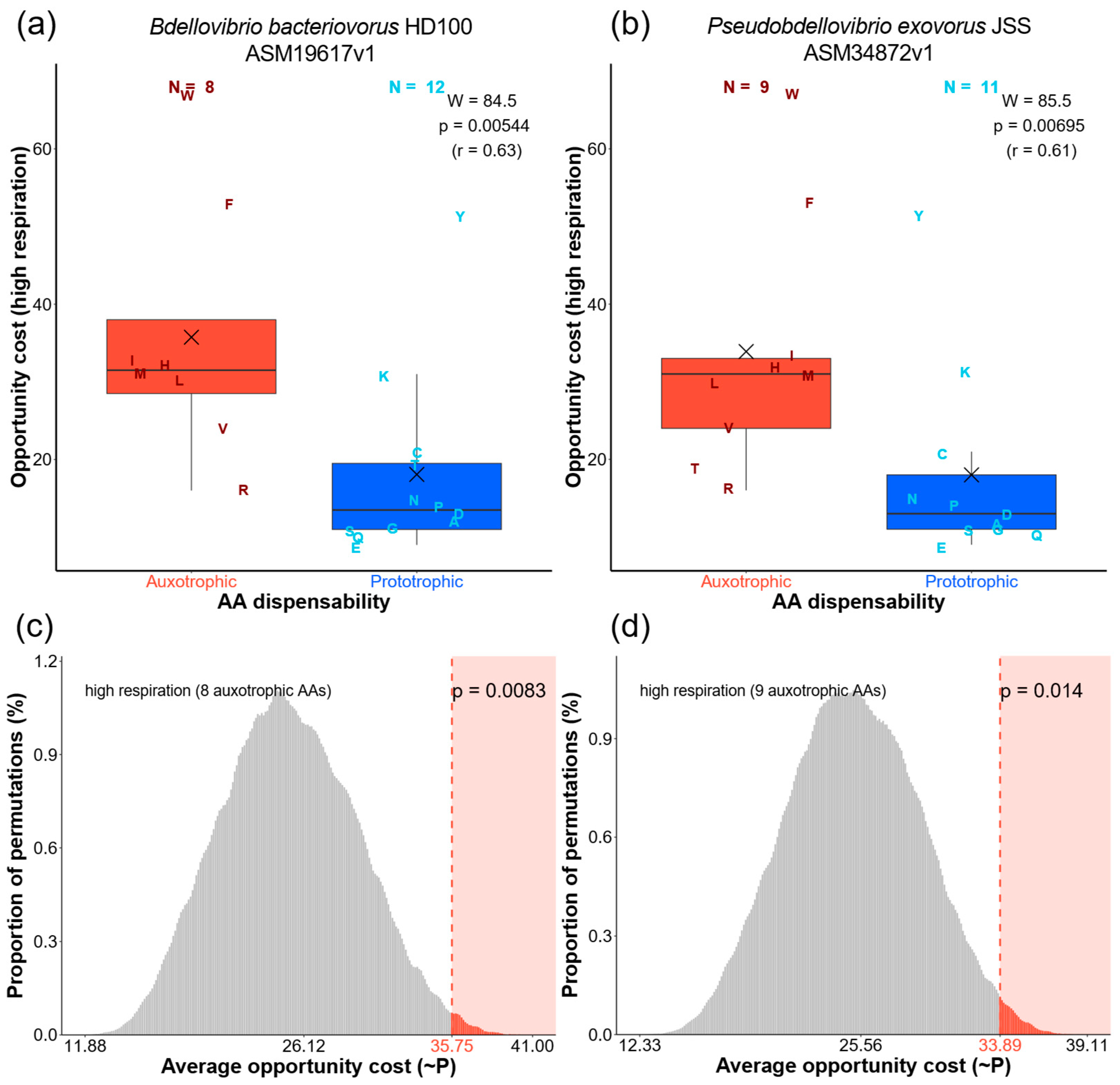
2.2. Expensive AAs Are Commonly Outsourced in Bdellovibrionota
2.3. Energy-Optimizing Selection Drives AA Auxotrophies in Bdellovibrionota
2.4. Bdellovibrionota Encode Expensive Proteomes
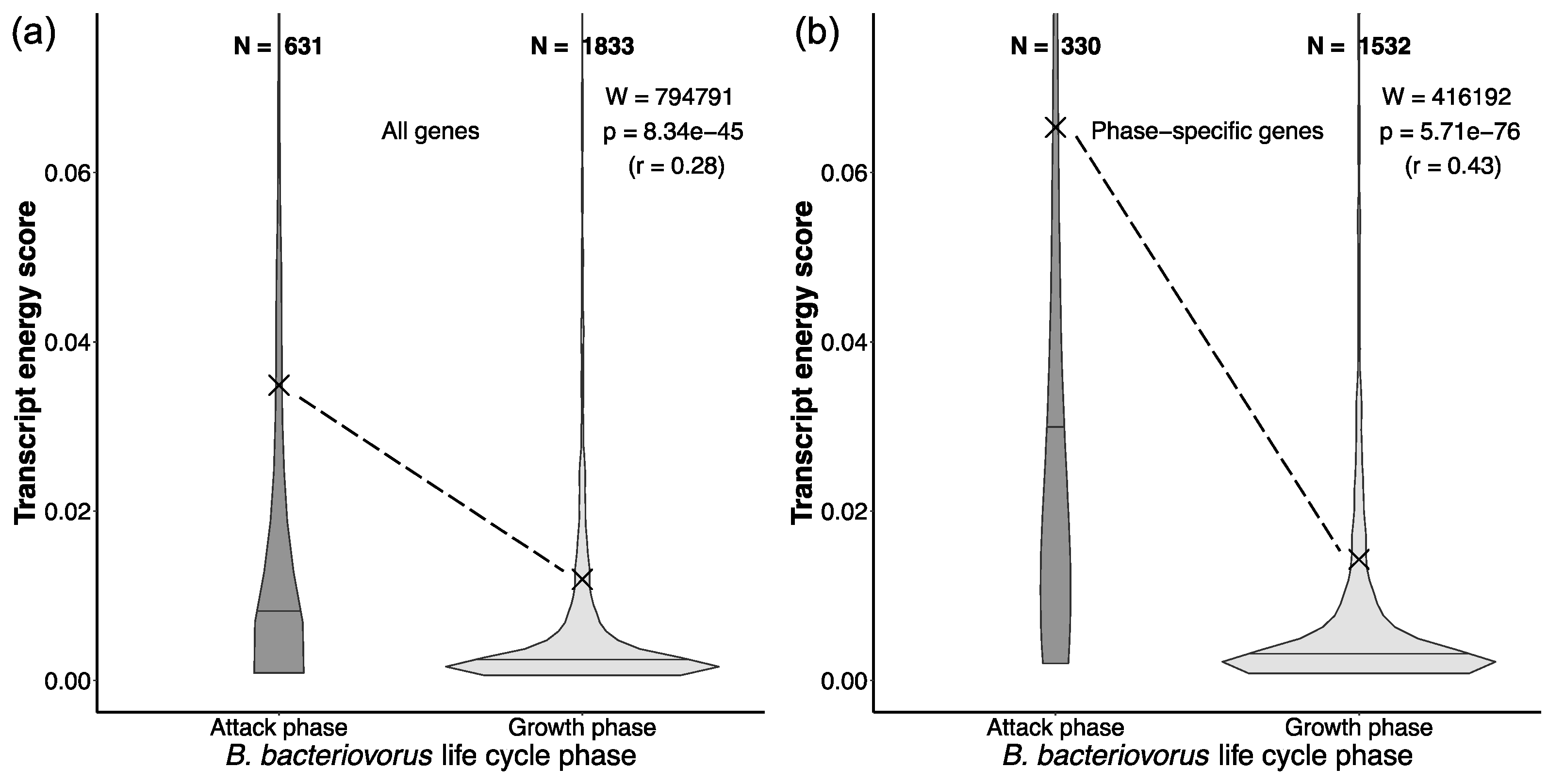
2.5. Expensive Proteins in Bdellovibrio Drive Active Predation
2.6. Expensive Proteins Are Functionally Understudied
3. Discussion
4. Materials and Methods
4.1. Databases, Completeness Score, and Auxotrophy Index
4.2. Opportunity Cost Measures, Permutation and Transcriptome Analyses
4.3. COG Functions Enrichment Analyses
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notations and Acronyms | Meaning | Equation |
|---|---|---|
| AA | Amino acid | - |
| AAs | Amino acids | - |
| AI | Auxotrophy index | - |
| AIi,j | Auxotrophy index of amino acid i in species j | Equation (1) |
| CS | Completeness score | - |
| CSi,j | Completeness score of amino acid i in species j | Equation (1) |
| Average auxotrophy index of amino acid i | Equation (1) | |
| OC | Opportunity cost | - |
| Opportunity cost of amino acid i | Equations (2) and (4) | |
| Protein opportunity cost | Equations (3)–(5) | |
| Number of occurrences of amino acid i in a protein | Equation (4) | |
| Frequency of amino acid i in a protein | Equation (4) | |
| Proteome opportunity cost | Equation (2) | |
| Number of occurrences of amino acid i in a proteome | Equation (2) | |
| Frequency of amino acid i in a proteome | Equation (2) | |
| TES | Transcript energy score | - |
| Transcript energy score of gene v | Equation (3) | |
| tv | Transcription value of gene v | Equation (3) |
| Transcription frequency of gene v in a transcriptome | Equation (3) | |
| Cluster opportunity cost | Equation (5) | |
| Number of proteins in a cluster | Equation (5) |
References
- Kasalo, N.; Domazet-Lošo, M.; Domazet-Lošo, T. Massive Outsourcing of Energetically Costly Amino Acids at the Origin of Animals. bioRxiv 2024. [Google Scholar] [CrossRef]
- Payne, S.H.; Loomis, W.F. Retention and Loss of Amino Acid Biosynthetic Pathways Based on Analysis of Whole-Genome Sequences. Eukaryot. Cell 2006, 5, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Trolle, J.; McBee, R.M.; Kaufman, A.; Pinglay, S.; Berger, H.; German, S.; Liu, L.; Shen, M.J.; Guo, X.; Martin, J.A.; et al. Resurrecting Essential Amino Acid Biosynthesis in Mammalian Cells. eLife 2022, 11, e72847. [Google Scholar] [CrossRef]
- Ramoneda, J.; Jensen, T.B.N.; Price, M.N.; Casamayor, E.O.; Fierer, N. Taxonomic and Environmental Distribution of Bacterial Amino Acid Auxotrophies. Nat. Commun. 2023, 14, 7608. [Google Scholar] [CrossRef]
- Kasalo, N.; Domazet-Lošo, T.; Domazet-Lošo, M. Bacterial Amino Acid Auxotrophies Enable Energetically Costlier Proteomes. Int. J. Mol. Sci. 2025, 26, 2285. [Google Scholar] [CrossRef]
- Starke, S.; Harris, D.M.M.; Zimmermann, J.; Schuchardt, S.; Oumari, M.; Frank, D.; Bang, C.; Rosenstiel, P.; Schreiber, S.; Frey, N.; et al. Amino Acid Auxotrophies in Human Gut Bacteria Are Linked to Higher Microbiome Diversity and Long-Term Stability. ISME J. 2023, 17, 2370–2380. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Waschina, S.; Pande, S.; Bohl, K.; Kaleta, C.; Kost, C. Less Is More: Selective Advantages CAN Explain the Prevalent Loss of Biosynthetic Genes in Bacteria. Evolution 2014, 68, 2559–2570. [Google Scholar] [CrossRef]
- Domazet-Lošo, M.; Široki, T.; Šimičević, K.; Domazet-Lošo, T. Macroevolutionary Dynamics of Gene Family Gain and Loss along Multicellular Eukaryotic Lineages. Nat. Commun. 2024, 15, 2663. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Knight, R. Global Patterns in Bacterial Diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef]
- Price, M.N.; Zane, G.M.; Kuehl, J.V.; Melnyk, R.A.; Wall, J.D.; Deutschbauer, A.M.; Arkin, A.P. Filling Gaps in Bacterial Amino Acid Biosynthesis Pathways with High-Throughput Genetics. PLoS Genet. 2018, 14, e1007147. [Google Scholar] [CrossRef]
- Sockett, R.E. Predatory Lifestyle of Bdellovibrio bacteriovorus. Annu. Rev. Microbiol. 2009, 63, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kadouri, D.E.; Wu, M. Genomic Insights into an Obligate Epibiotic Bacterial Predator: Micavibrio aeruginosavorus ARL-13. BMC Genom. 2011, 12, 453. [Google Scholar] [CrossRef]
- Ye, X.-S.; Chen, M.-X.; Li, H.-Y.; He, X.-Y.; Zhao, Y. Halobacteriovorax vibrionivorans Sp. Nov., a Novel Prokaryotic Predator Isolated from Coastal Seawater of China. Int. J. Syst. Evol. Microbiol. 2019, 69, 3917–3923. [Google Scholar] [CrossRef]
- Karunker, I.; Rotem, O.; Dori-Bachash, M.; Jurkevitch, E.; Sorek, R. A Global Transcriptional Switch between the Attack and Growth Forms of Bdellovibrio bacteriovorus. PLoS ONE 2013, 8, e61850. [Google Scholar] [CrossRef]
- Davis, S.C.; Cerra, J.; Williams, L.E. Comparative Genomics of Obligate Predatory Bacteria Belonging to Phylum Bdellovibrionota Highlights Distribution and Predicted Functions of Lineage-Specific Protein Families. mSphere 2024, 9, e00680-24. [Google Scholar] [CrossRef]
- Waite, D.W.; Chuvochina, M.; Pelikan, C.; Parks, D.H.; Yilmaz, P.; Wagner, M.; Loy, A.; Naganuma, T.; Nakai, R.; Whitman, W.B.; et al. Proposal to Reclassify the Proteobacterial Classes Deltaproteobacteria and Oligoflexia, and the Phylum Thermodesulfobacteria into Four Phyla Reflecting Major Functional Capabilities. Int. J. Syst. Evol. Microbiol. 2020, 70, 5972–6016. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Dey, A.; Vassallo, C.N.; Wall, D. How Myxobacteria Cooperate. J. Mol. Biol. 2015, 427, 3709–3721. [Google Scholar] [CrossRef] [PubMed]
- Hungate, B.A.; Marks, J.C.; Power, M.E.; Schwartz, E.; Van Groenigen, K.J.; Blazewicz, S.J.; Chuckran, P.; Dijkstra, P.; Finley, B.K.; Firestone, M.K.; et al. The Functional Significance of Bacterial Predators. mBio 2021, 12, e00466-21. [Google Scholar] [CrossRef]
- Kaiser, D.; Robinson, M.; Kroos, L. Myxobacteria, Polarity, and Multicellular Morphogenesis. Cold Spring Harb. Perspect. Biol. 2010, 2, a000380. [Google Scholar] [CrossRef]
- Reichenbach, H. The Ecology of the Myxobacteria. Environ. Microbiol. 1999, 1, 15–21. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. A Phylogenetically Based Transcriptome Age Index Mirrors Ontogenetic Divergence Patterns. Nature 2010, 468, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, X.; Zhou, J.; Ju, F. Active Predation, Phylogenetic Diversity, and Global Prevalence of Myxobacteria in Wastewater Treatment Plants. ISME J. 2023, 17, 671–681. [Google Scholar] [CrossRef]
- Muñoz-Dorado, J.; Marcos-Torres, F.J.; García-Bravo, E.; Moraleda-Muñoz, A.; Pérez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef]
- Mee, M.T.; Collins, J.J.; Church, G.M.; Wang, H.H. Syntrophic Exchange in Synthetic Microbial Communities. Proc. Natl. Acad. Sci. USA 2014, 111, E2149–E2156. [Google Scholar] [CrossRef]
- Shoguchi, E. Gene Clusters for Biosynthesis of Mycosporine-like Amino Acids in Dinoflagellate Nuclear Genomes: Possible Recent Horizontal Gene Transfer between Species of Symbiodiniaceae (Dinophyceae). J. Phycol. 2022, 58, 1–11. [Google Scholar] [CrossRef]
- Sockett, R.E.; Lambert, C. Bdellovibrio as Therapeutic Agents: A Predatory Renaissance? Nat. Rev. Microbiol. 2004, 2, 669–675. [Google Scholar] [CrossRef]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. SciPy 2010, 7, 92–96. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasalo, N.; Domazet-Lošo, M.; Domazet-Lošo, T. Convergence in Amino Acid Outsourcing Between Animals and Predatory Bacteria. Int. J. Mol. Sci. 2025, 26, 3024. https://doi.org/10.3390/ijms26073024
Kasalo N, Domazet-Lošo M, Domazet-Lošo T. Convergence in Amino Acid Outsourcing Between Animals and Predatory Bacteria. International Journal of Molecular Sciences. 2025; 26(7):3024. https://doi.org/10.3390/ijms26073024
Chicago/Turabian StyleKasalo, Niko, Mirjana Domazet-Lošo, and Tomislav Domazet-Lošo. 2025. "Convergence in Amino Acid Outsourcing Between Animals and Predatory Bacteria" International Journal of Molecular Sciences 26, no. 7: 3024. https://doi.org/10.3390/ijms26073024
APA StyleKasalo, N., Domazet-Lošo, M., & Domazet-Lošo, T. (2025). Convergence in Amino Acid Outsourcing Between Animals and Predatory Bacteria. International Journal of Molecular Sciences, 26(7), 3024. https://doi.org/10.3390/ijms26073024