
From DNA Repair to Redox Signaling: The Multifaceted Role of APEX1 (Apurinic/Apyrimidinic Endonuclease 1) in Cardiovascular Health and Disease

 ,
,
Abstract
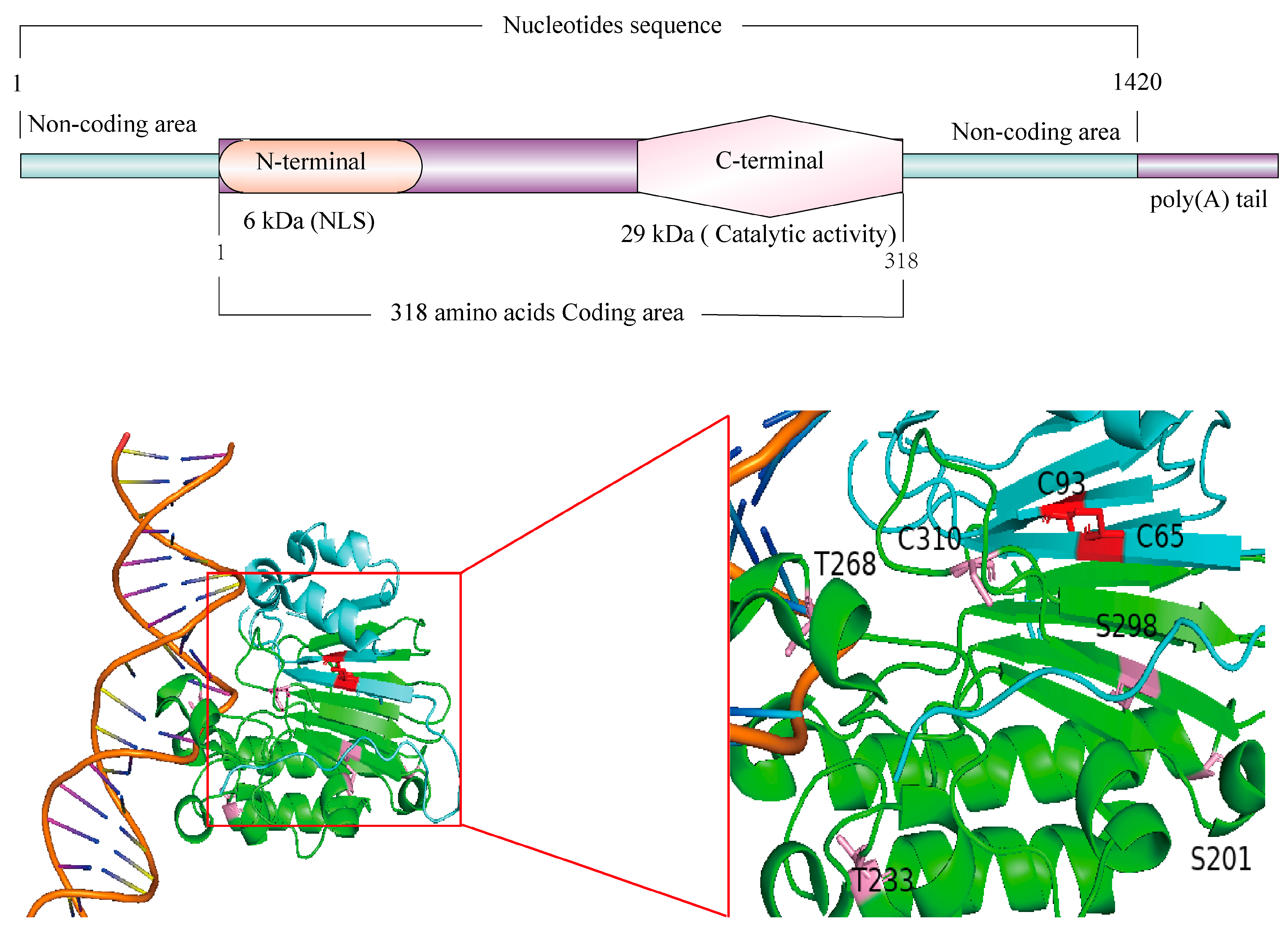
1. Discovery and Structure of APEX1
2. Functions of APEX1
2.1. N-Terminal Domain DNA/RNA/Protein Binding and Redox Activity
2.2. C-Terminal Domain Endonuclease Activity
2.3. 3′-Exonuclease Activity
2.4. Apex1 in Cardiovascular Physiology
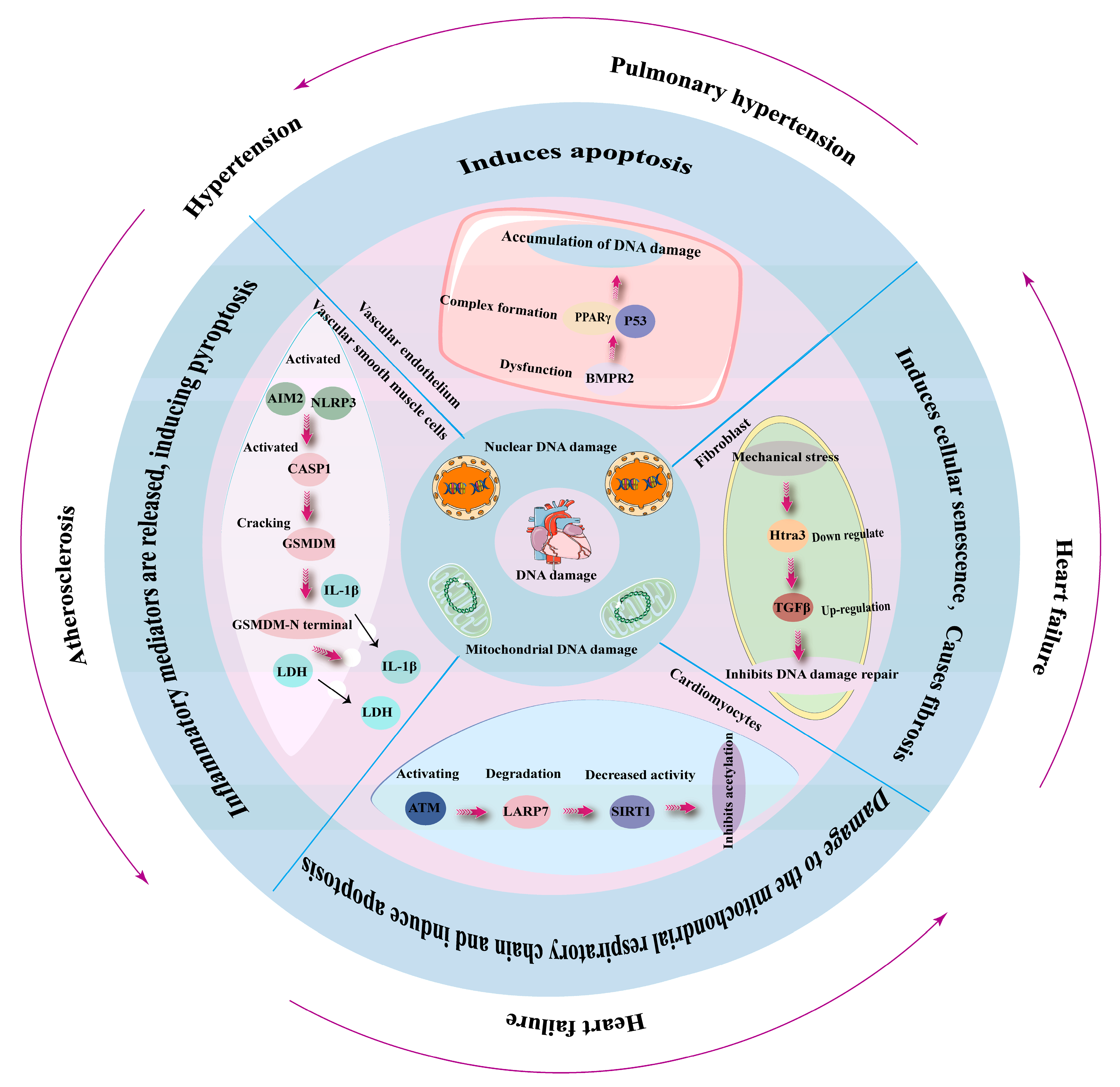
2.5. DNA Damage and Diseases of the Cardiovascular System
3. APEX1 in Early Embryonic Development, Aging and Diseases
4. APEX1 in Cardiovascular Cells and Diseases
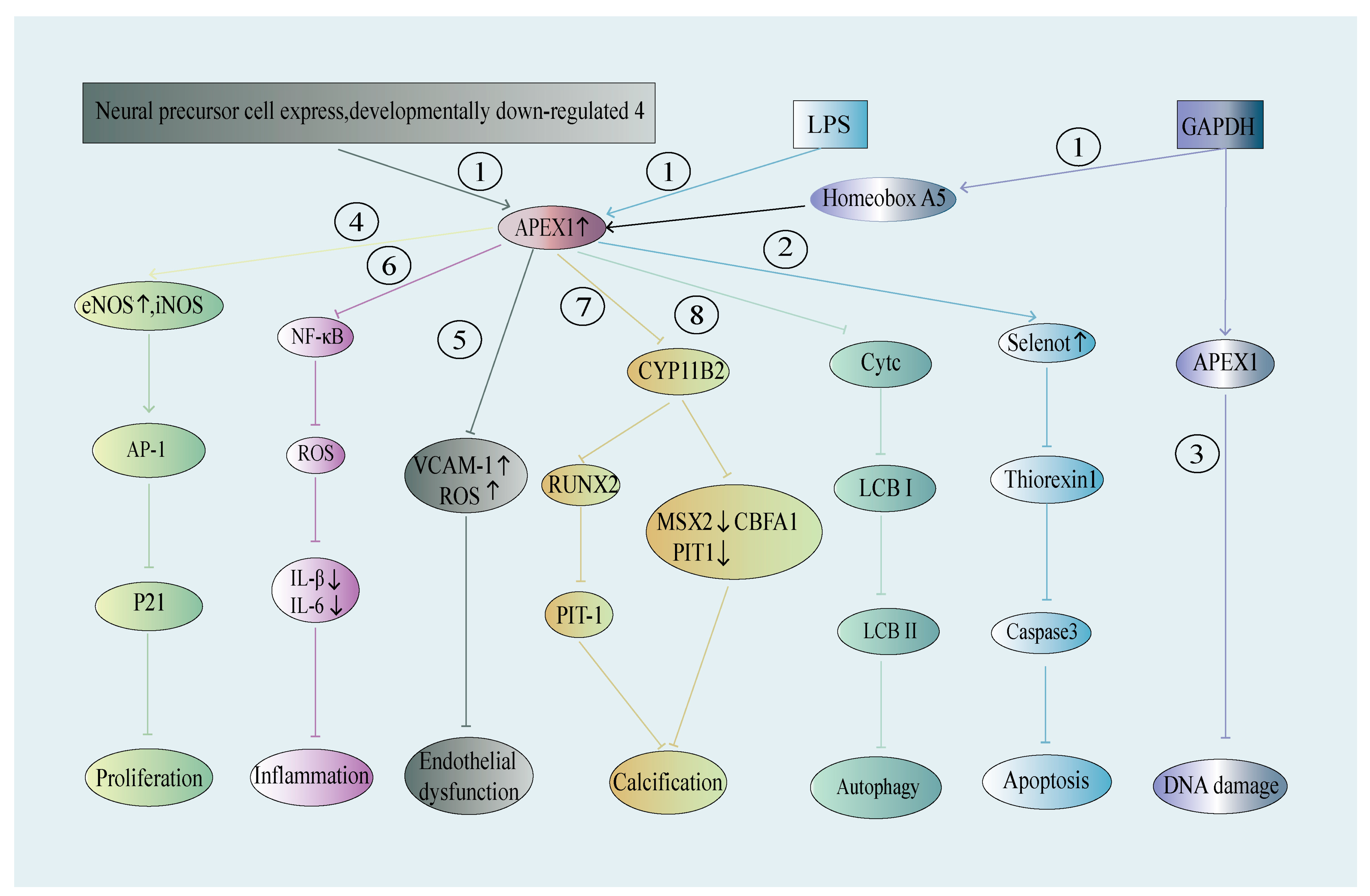
4.1. APEX1 in ECs—Its Role in Hypertension, Atherosclerosis, and Other Vascular Diseases
4.1.1. APEX1 in ECs and Hypertension
4.1.2. APEX1 in ECs and Atherosclerosis
4.1.3. APEX1 and Stroke—Potential Involvement of EC APEX1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type/Tissue | Agents/Models/Samples | Expression Levels | Effects of APEX1 Expression | |
|---|---|---|---|---|
| Cardiomyocytes | H9c2/Hips-CMs cells | Oxidative stress (H2O2) | Protein ↓ | Involved in apoptosis via caspase 3 pathway [156] |
| Heart samples (mice) | Transverse aortic constriction (TCA) | WT TCA: Protein ↓ Dec1 KO TAC: Protein ↓ Dec1 KO: Protein → | N/A [157] | |
| Neonatal cardiomyocyte (rat) | Miconazole-stimulated | mRNA ↓, Protein ↓ | Cell apoptosis [158] | |
| H9c2 (rat) | Hypoxia/reoxygenation (H/R) | Protein ↑ | Overexpression of APE1 attenuates cardiac H/R injury and promotes PINK1/Parkin-mediated mitophagy in H/R-injured H9c2 cells [159] | |
| Endothelial Cells (ECs) | HUVECs | Oxidized low-density lipoprotein | Protein ↓ | Inflammation and dysfunction [146] |
| MS-1 (CRL-2279™) (mice) | phorbol 12-myristate 13-acetate (PMA) | Mitochondrial: Protein ↑ Nucleus: Protein ↓ | Mitochondrial dysfunction [147] | |
| HUVEC, HEK 293, liver/kidney/ aorta samples of mice | Oxidative stress (H2O2) APE1/Ref-1+/− mice | Protein ↓ | Endothelial SIRT1 inactivation [128] | |
| BPAEC/HULEC-5a | 17-AAG, 17-DMAG, AUY-922 | Protein ↓ | Enhances the barrier effect in HULEC-5a and BPAEC [160] | |
| HUVECs, CPAEs | Hypoxic injury, TNF-α | Protein ↓ | HUVECs and CPAEs apoptosis [50] | |
| HUVECs | TNF-α | Protein ↓ | Monocyte adhesion in ECs [143] | |
| Primary human ECs | Oxidative stress (H2O2) | Protein ↓ | Apoptosis [161] | |
| Vascular Smooth Muscle Cells (SMCs) | Vascular SMCs (rat) | Phosphate-induced | Protein ↓ | Apoptosis, calcification, and osteoblastic phenotype changes in vascular SMCs [117] |
| Rat aortic SMCs | H2O2 | Protein ↓ | DNA damage, apoptosis and cell death [115] | |
| Vascular SMCs (mice) | iNOS KO | N/A | Altered AP-1/Ref-1 redox pathway and reduced proliferative response [114] | |
| RASMC, Rat carotid arteries | Angiotensin II, Balloon-injured | Nuclear fraction: Protein ↑ Cytosolic fraction: Protein ↓ Total → | Neointimal formation and vascular SMC migration [162] | |
4.2. APEX1 in Vascular SMCs—A Role in Arterial Neointima and Calcification
4.3. APEX1 in Cardiomyocytes—A Role in Myocardial Ischemia Injury
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| AIM | absent in melanoma |
| AIM2 | absent in melanoma 2 |
| Ang II | angiotensin II |
| AP | apyrimidinic |
| AP-1 | activator protein 1 |
| APEX1 | apurinic/apyrimidinarric endonuclease 1 |
| AP-site | apyrimidinic site |
| AS | atherosclerosis |
| BER | base excision repair |
| BMPR2 | bone morphogenetic protein receptor type 2 |
| BPAEC | bovine pulmonary arterial endothelial cells |
| CASP1 | caspase 1 |
| CVD | cardiovascular diseases |
| Cytc | cytochrome c |
| DNA-pol | DNA polymerase |
| DSR-Pathway | double-strand break repair pathway |
| EC | endothelial cell |
| eNOS | endothelial NO synthase |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular regulated protein kinases 1/2 |
| ESC | embryonic stem cell |
| Exo III | endonuclease III |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GDNF | glial cell derived neurotrophic factor |
| GLP-1 | glucagon-like peptide-1 |
| GSEA | gene enrichment analysis |
| GSMDM | gasdermin |
| GSMDM N-terminal | N-terminal of GSDMD |
| H/R | hypoxia/reoxygenation |
| HDAC | histone deacetylase |
| HEK 293 | human embryonic kidney cells |
| HF | Heart Failure |
| HULEC-5a | human lung microvascular endothelium cells |
| HUVECs | human umbilical vein endothelial cells |
| IL-1β | interleukin-1 beta |
| IL-6 | interleukin-6 |
| iNOS | inducible nitric oxide synthase |
| Htra3 | htra serine peptidase 3 |
| KO | knockout |
| LARP7 | la ribonucleoprotein 7 |
| LCBⅠ | microtubule associated protein 1 light chain 3 alpha |
| LCBⅡ | microtubule associated protein 1 light chain 3 beta |
| LDH | lactate dehydrogenase |
| LPS | lipopolysaccharide |
| mt-DNA | mitochondrial DNA |
| NADPH | nicotinamide-adenine dinucleotide phosphate |
| NEDD4 | neuronally expressed developmentally downregulated 4 |
| NF-κB | nuclear factor kappa-B |
| NLS | nuclear localization signal |
| NO | nitric oxide |
| NPM1 | nucleophosmin1 |
| oxLDL | oxidized low-density lipoprotein |
| P21 | cyclin dependent kinase inhibitor 1A |
| P53 | tumor protein P53 |
| PARP-1 | poly (ADP-ribose) polymerase 1 |
| PIT1 | pituitary-specific positive transcription factor |
| PINK1 | PTEN induced putative kinase 1 |
| PKC | protein kinase C |
| PMA | propidium monoazidos |
| Pol | polymerase |
| PPAR-γ | peroxisome proliferator activated receptor γ |
| PPI | protein-protein interaction |
| PTM | protein translational modification |
| ROS | reactive oxygen species |
| ROS | reactive oxygen species |
| RUNX2 | runt-related transcription factor 2 |
| SIRT1 | silent mating type information regulation 2 |
| SNP | single nucleotide polymorphism |
| ss-DNA | single-stranded DNA |
| SSR-Pathway | single-strand break repair pathway |
| STAT3 | signal transducer and activator of transcription 3 |
| TGFβ | transforming growth factor beta |
| TNF | tumor necrosis factor |
| VCAM | vascular cell adhesion molecule |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VEGF | vascular endothelial growth factor |
| VSMC | vascular smooth muscle cell |
| WT | wide type |
| XRCC1 | X-ray repair cross-complementing 1 |
| 17-DMAG | alvespimycin |
| 17-AGG | tanespimycin |
| AUY-922 | luminespib |
References
- Malfatti, M.C.; Bellina, A.; Antoniali, G.; Tell, G. Revisiting Two Decades of Research Focused on Targeting APE1 for Cancer Therapy: The Pros and Cons. Cells 2023, 12, 1859. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Linn, S. On the recognition and cleavage mechanism of Escherichia coli endodeoxyribonuclease V, a possible DNA repair enzyme. J. Biol. Chem. 1982, 257, 2848–2855. [Google Scholar] [PubMed]
- Linsley, W.S.; Penhoet, E.E.; Linn, S. Human endonuclease specific for apurinic/apyrimidinic sites in DNA. Partial purification and characterization of mul-tiple forms from placenta. J. Biol. Chem. 1977, 252, 1235–1242. [Google Scholar]
- Wilson, D.M., 3rd; Bennett, R.A.; Marquis, J.C.; Ansari, P.; Demple, B. Trans-complementation by human apurinic endonuclease (Ape) of hypersensitivity to DNA damage and spontaneous mutator phenotype in apn1-yeast. Nucleic Acids Res. 1995, 23, 5027–5033. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Takeshita, M.; Demple, B. Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res. 1997, 25, 933–939. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Curran, T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992, 11, 653–665. [Google Scholar] [CrossRef]
- Spiro, I.J.; Barrows, L.R.; Kennedy, K.A.; Ling, C.C. Transfection of a human gene for the repair of X-ray- and EMS-induced DNA damage. Radiat. Res. 1986, 108, 146–157. [Google Scholar]
- Seki, S.; Hatsushika, M.; Watanabe, S.; Akiyama, K.; Nagao, K.; Tsutsui, K. cDNA cloning, sequencing, expression and possible domain structure of human APEX nuclease homologous to Escherichia coli exonuclease III. Biochim. Biophys. Acta 1992, 1131, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Mosbaugh, D.W.; Linn, S. Further characterization of human fibroblast apurinic/apyrimidinic DNA endonucleases. The definition of two mechanistic classes of enzyme. J. Biol. Chem. 1980, 255, 11743–11752. [Google Scholar]
- Robson, C.N.; Hochhauser, D.; Craig, R.; Rack, K.; Buckle, V.J.; Hickson, I.D. Structure of the human DNA repair gene HAP1 and its localisation to chromosome 14q 11.2-12. Nucleic Acids Res. 1992, 20, 4417–4421. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Hang, B.; Gorman, M.A.; Freemont, P.S.; Singer, B.; Hickson, I.D. Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilises substrate binding. Nucleic Acids Res. 2000, 28, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Grandy, D.K.; Hagerup, J.M.; Magenis, R.E.; Smith, L.; Chauhan, B.C.; Henner, W.D. The human gene for apurinic/apyrimidinic endonuclease (HAP1): Sequence and localization to chromosome 14 band q12. Nucleic Acids Res. 1992, 20, 4097–4098. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected]. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Beard, W.A.; Cuneo, M.J.; Dyrkheeva, N.S.; Wilson, S.H. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015, 22, 924–931. [Google Scholar] [CrossRef]
- Beloglazova, N.G.; Kirpota, O.O.; Starostin, K.V.; Ishchenko, A.A.; Yamkovoy, V.I.; Zharkov, D.O.; Douglas, K.T.; Nevinsky, G.A. Thermodynamic, kinetic and structural basis for recognition and repair of abasic sites in DNA by apurinic/apyrimidinic endonuclease from human placenta. Nucleic Acids Res. 2004, 32, 5134–5146. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kong, M.; Gassman, N.R.; Freudenthal, B.D.; Prasad, R.; Zhen, S.; Watkins, S.C.; Wilson, S.H.; Van Houten, B. PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1. Nucleic Acids Res. 2017, 45, 12834–12847. [Google Scholar] [CrossRef]
- Strauss, P.R.; Beard, W.A.; Patterson, T.A.; Wilson, S.H. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997, 272, 1302–1307. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.G.; Curran, T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA 1994, 91, 23–27. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef]
- Izumi, T.; Mitra, S. Deletion analysis of human AP-endonuclease: Minimum sequence required for the endonuclease activity. Carcinogenesis 1998, 19, 525–527. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef]
- Jackson, E.B.; Theriot, C.A.; Chattopadhyay, R.; Mitra, S.; Izumi, T. Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1). Nucleic Acids Res. 2005, 33, 3303–3312. [Google Scholar] [CrossRef]
- Fantini, D.; Vascotto, C.; Marasco, D.; D’Ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F.; et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef] [PubMed]
- Antoniali, G.; Serra, F.; Lirussi, L.; Tanaka, M.; D’Ambrosio, C.; Zhang, S.; Radovic, S.; Dalla, E.; Ciani, Y.; Scaloni, A.; et al. Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism. Nat. Commun. 2017, 8, 797. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.M.; Stark, W.J.; Flynn, T.S.; Freudenthal, B.D. Molecular and structural characterization of disease-associated APE1 polymorphisms. DNA Repair 2020, 91-92, 102867. [Google Scholar] [CrossRef] [PubMed]
- Howpay Manage, S.A.; Zhu, J.; Fleming, A.M.; Burrows, C.J. Promoters vs. telomeres: AP-endonuclease 1 interactions with abasic sites in G-quadruplex folds depend on topology. RSC Chem. Biol. 2023, 4, 261–270. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 4039–4054. [Google Scholar] [CrossRef]
- Antoniali, G.; Lirussi, L.; D’Ambrosio, C.; Dal Piaz, F.; Vascotto, C.; Casarano, E.; Marasco, D.; Scaloni, A.; Fogolari, F.; Tell, G. SIRT1 gene expression upon genotoxic damage is regulated by APE1 through nCaRE-promoter elements. Mol. Biol. Cell 2014, 25, 532–547. [Google Scholar] [CrossRef]
- Erzberger, J.P.; Wilson, D.M., 3rd. The role of Mg2+ and specific amino acid residues in the catalytic reaction of the major human abasic endonuclease: New insights from EDTA-resistant incision of acyclic abasic site analogs and site-directed mutagenesis. J. Mol. Biol. 1999, 290, 447–457. [Google Scholar] [CrossRef]
- Fleming, A.M.; Manage, S.A.H.; Burrows, C.J. Binding of AP endonuclease-1 to G-quadruplex DNA depends on the N-terminal domain, Mg(2+) and ionic strength. ACS Bio. Med. Chem. Au J. 2021, 1, 44–56. [Google Scholar] [CrossRef]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA charge transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Berquist, B.R.; McNeill, D.R.; Wilson, D.M., 3rd. Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J. Mol. Biol. 2008, 379, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.A.; Morera, S.; Rothwell, D.G.; de La Fortelle, E.; Mol, C.D.; Tainer, J.A.; Hickson, I.D.; Freemont, P.S. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997, 16, 6548–6558. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Freudenthal, B.D. APE1: A skilled nucleic acid surgeon. DNA Repair 2018, 71, 93–100. [Google Scholar] [CrossRef]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: From mechanism to disease. Frontiers in Bioscience (Landmark Ed.) 2017, 22, 1493–1522. [Google Scholar] [CrossRef]
- Wilson, S.H.; Kunkel, T.A. Passing the baton in base excision repair. Nat. Struct. Biol. 2000, 7, 176–178. [Google Scholar] [CrossRef]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair 2007, 6, 317–328. [Google Scholar] [CrossRef]
- Kladova, O.A.; Bazlekowa-Karaban, M.; Baconnais, S.; Pietrement, O.; Ishchenko, A.A.; Matkarimov, B.T.; Iakovlev, D.A.; Vasenko, A.; Fedorova, O.S.; Le Cam, E.; et al. The role of the N-terminal domain of human apurinic/apyrimidinic endonuclease 1, APE1, in DNA glycosylase stimulation. DNA Repair 2018, 64, 10–25. [Google Scholar] [CrossRef]
- Maher, R.L.; Bloom, L.B. Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J. Biol. Chem. 2007, 282, 30577–30585. [Google Scholar] [CrossRef]
- Li, M.; Zhong, Z.; Zhu, J.; Xiang, D.; Dai, N.; Cao, X.; Qing, Y.; Yang, Z.; Xie, J.; Li, Z.; et al. Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J. Biol. Chem. 2010, 285, 14871–14881. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, J.; He, H.; Su, D.; Chen, Q.; Gross, M.L.; Kelley, M.R.; Georgiadis, M.M. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry 2012, 51, 695–705. [Google Scholar] [CrossRef]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. NPJ Precis. Oncol. 2017, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y.; Yu, Z.; Zyromski, N.; Schipani, E.; Carta, F.; et al. Regulation of HIF1alpha under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual Targeting in Patient-Derived 3D Pancreatic Cancer Models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.L.; Jiang, Y.; Rajeshkumar, N.V.; Scandura, G.; Sinn, A.L.; He, Y.; Shen, C.; Jones, D.R.; Pollok, K.E.; Ivan, M.; et al. Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol. Cancer Ther. 2011, 10, 1698–1708. [Google Scholar] [CrossRef]
- Oliveira, T.T.; Coutinho, L.G.; de Oliveira, L.O.A.; Timoteo, A.R.S.; Farias, G.C.; Agnez-Lima, L.F. APE1/Ref-1 Role in Inflammation and Immune Response. Front. Immunol. 2022, 13, 793096. [Google Scholar] [CrossRef]
- Jedinak, A.; Dudhgaonkar, S.; Kelley, M.R.; Sliva, D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011, 31, 379–385. [Google Scholar]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Hall, J.L.; Wang, X.; Van, A.; Zhao, Y.; Gibbons, G.H. Overexpression of Ref-1 inhibits hypoxia and tumor necrosis factor-induced endothelial cell apoptosis through nuclear factor-kappab-independent and -dependent pathways. Circ. Res. 2001, 88, 1247–1253. [Google Scholar] [CrossRef]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef]
- Ramana, C.V.; Boldogh, I.; Izumi, T.; Mitra, S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc. Natl. Acad. Sci. USA 1998, 95, 5061–5066. [Google Scholar] [CrossRef]
- Jayaraman, L.; Murthy, K.G.; Zhu, C.; Curran, T.; Xanthoudakis, S.; Prives, C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997, 11, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, O.I.; Prasad, R.; Sobol, R.W.; Horton, J.K.; Ackerman, E.J.; Wilson, S.H. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem. 2001, 276, 25541–25548. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Bennett, R.A.; Demple, B. Dynamics of the interaction of human apurinic endonuclease (Ape1) with its substrate and product. J. Biol. Chem. 1998, 273, 30352–30359. [Google Scholar] [CrossRef]
- Giorgio, M.; Dellino, G.I.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef]
- Chou, K.M.; Cheng, Y.C. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3’ mispaired DNA. Nature 2002, 415, 655–659. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Khodyreva, S.N.; Sukhanova, M.V.; Safronov, I.V.; Dezhurov, S.V.; Lavrik, O.I. 3’-5’ exonuclease activity of human apurinic/apyrimidinic endonuclease 1 towards DNAs containing dNMP and their modified analogs at the 3 end of single strand DNA break. Biochemistry 2006, 71, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Dyrkheeva, N.S.; Lomzov, A.A.; Pyshnyi, D.V.; Khodyreva, S.N.; Lavrik, O.I. Efficiency of exonucleolytic action of apurinic/apyrimidinic endonuclease 1 towards matched and mismatched dNMP at the 3’ terminus of different oligomeric DNA structures correlates with thermal stability of DNA duplexes. Biochim. Biophys. Acta 2006, 1764, 699–706. [Google Scholar] [CrossRef]
- Hadi, M.Z.; Ginalski, K.; Nguyen, L.H.; Wilson, D.M., 3rd. Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J. Mol. Biol. 2002, 316, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Hoitsma, N.M.; Whitaker, A.M.; Beckwitt, E.C.; Jang, S.; Agarwal, P.K.; Van Houten, B.; Freudenthal, B.D. AP-endonuclease 1 sculpts DNA through an anchoring tyrosine residue on the DNA intercalating loop. Nucleic Acids Res. 2020, 48, 7345–7355. [Google Scholar] [CrossRef]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.A.; Hashiguchi, K.; Wilson, D.M., 3rd; Copeland, W.C.; Souza-Pinto, N.C.; Bohr, V.A. DNA base excision repair activities and pathway function in mitochondrial and cellular lysates from cells lacking mitochondrial DNA. Nucleic Acids Res. 2004, 32, 2181–2192. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef]
- Matsushita, H.; Morishita, R.; Nata, T.; Aoki, M.; Nakagami, H.; Taniyama, Y.; Yamamoto, K.; Higaki, J.; Yasufumi, K.; Ogihara, T. Hypoxia-induced endothelial apoptosis through nuclear factor-kappaB (NF-kappaB)-mediated bcl-2 suppression: In vivo evidence of the importance of NF-kappaB in endothelial cell regulation. Circ. Res. 2000, 86, 974–981. [Google Scholar] [CrossRef]
- Angkeow, P.; Deshpande, S.S.; Qi, B.; Liu, Y.X.; Park, Y.C.; Jeon, B.H.; Ozaki, M.; Irani, K. Redox factor-1: An extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Differ. 2002, 9, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef]
- Nakada, Y.; Nhi Nguyen, N.U.; Xiao, F.; Savla, J.J.; Lam, N.T.; Abdisalaam, S.; Bhattacharya, S.; Mukherjee, S.; Asaithamby, A.; Gillette, T.G.; et al. DNA Damage Response Mediates Pressure Overload-Induced Cardiomyocyte Hypertrophy. Circulation 2019, 139, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Drake, K.M.; Rigelsky, C.M.; McNelly, L.N.; Meade, S.L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Ide, T.; Shiomi, T.; Kang, D.; Hayashidani, S.; Suematsu, N.; Wen, J.; Utsumi, H.; Hamasaki, N.; Takeshita, A. 8-oxo-dGTPase, which prevents oxidative stress-induced DNA damage, increases in the mitochondria from failing hearts. Circulation 2001, 104, 2883–2885. [Google Scholar] [CrossRef]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell. 2008, 30, 277–289. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; Van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res. 2000, 86, 960–966. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Mercer, J.; Bennett, M. DNA damage and repair in atherosclerosis. Cardiovasc. Res. 2006, 71, 259–268. [Google Scholar] [CrossRef]
- Martinet, W.; Knaapen, M.W.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002, 106, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Q.; Wang, T.; Long, Q.; Sun, Y.; Jiao, L.; Gullerova, M. DNA damage response, a double-edged sword for vascular aging. Ageing Res. Rev. 2023, 92, 102137. [Google Scholar] [CrossRef]
- Higo, T.; Naito, A.T.; Sumida, T.; Shibamoto, M.; Okada, K.; Nomura, S.; Nakagawa, A.; Yamaguchi, T.; Sakai, T.; Hashimoto, A.; et al. DNA single-strand break-induced DNA damage response causes heart failure. Nat. Commun. 2017, 8, 15104. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sowers, J.R.; Zhang, Y.; Ren, J. Targeting DNA damage response in cardiovascular diseases: From pathophysiology to therapeutic implications. Cardiovasc. Res. 2023, 119, 691–709. [Google Scholar] [CrossRef]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Min, W.; Bradley, J.R. Mechanisms of endothelial dysfunction, injury, and death. Annu. Rev. Pathol. 2009, 4, 71–95. [Google Scholar] [CrossRef]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef]
- Mitomo, K.; Nakayama, K.; Fujimoto, K.; Sun, X.; Seki, S.; Yamamoto, K. Two different cellular redox systems regulate the DNA-binding activity of the p50 subunit of NF-kappa B in vitro. Gene 1994, 145, 197–203. [Google Scholar] [CrossRef]
- Nishi, T.; Shimizu, N.; Hiramoto, M.; Sato, I.; Yamaguchi, Y.; Hasegawa, M.; Aizawa, S.; Tanaka, H.; Kataoka, K.; Watanabe, H.; et al. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J. Biol. Chem. 2002, 277, 44548–44556. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wagner, E.F. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. Dis. 2011, 70 (Suppl. 1), i109–i112. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Vidula, N.; Dubash, T.; Lawrence, M.S.; Simoneau, A.; Niemierko, A.; Blouch, E.; Nagy, B.; Roh, W.; Chirn, B.; Reeves, B.A.; et al. Identification of Somatically Acquired BRCA1/2 Mutations by cfDNA Analysis in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 4852–4862. [Google Scholar] [CrossRef]
- Raffoul, J.J.; Cabelof, D.C.; Nakamura, J.; Meira, L.B.; Friedberg, E.C.; Heydari, A.R. Apurinic/apyrimidinic endonuclease (APE/REF-1) haploinsufficient mice display tissue-specific differences in DNA polymerase beta-dependent base excision repair. J. Biol. Chem. 2004, 279, 18425–18433. [Google Scholar] [CrossRef] [PubMed]
- Meira, L.B.; Devaraj, S.; Kisby, G.E.; Burns, D.K.; Daniel, R.L.; Hammer, R.E.; Grundy, S.; Jialal, I.; Friedberg, E.C. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001, 61, 5552–5557. [Google Scholar]
- Ludwig, D.L.; MacInnes, M.A.; Takiguchi, Y.; Purtymun, P.E.; Henrie, M.; Flannery, M.; Meneses, J.; Pedersen, R.A.; Chen, D.J. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat. Res. 1998, 409, 17–29. [Google Scholar] [CrossRef]
- Jang, J.; Huh, Y.J.; Cho, H.J.; Lee, B.; Park, J.; Hwang, D.Y.; Kim, D.W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem. Cell. Rep. 2017, 9, 629–641. [Google Scholar] [CrossRef]
- Domenis, R.; Bergamin, N.; Gianfranceschi, G.; Vascotto, C.; Romanello, M.; Rigo, S.; Vagnarelli, G.; Faggiani, M.; Parodi, P.; Kelley, M.R.; et al. The redox function of APE1 is involved in the differentiation process of stem cells toward a neuronal cell fate. PLoS ONE 2014, 9, e89232. [Google Scholar] [CrossRef]
- Zou, G.M.; Luo, M.H.; Reed, A.; Kelley, M.R.; Yoder, M.C. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood 2007, 109, 1917–1922. [Google Scholar] [CrossRef]
- Desai, R.V.; Chen, X.; Martin, B.; Chaturvedi, S.; Hwang, D.W.; Li, W.; Yu, C.; Ding, S.; Thomson, M.; Singer, R.H.; et al. A DNA repair pathway can regulate transcriptional noise to promote cell fate transitions. Science 2021, 373. [Google Scholar] [CrossRef]
- Li, M.; Yang, X.; Lu, X.; Dai, N.; Zhang, S.; Cheng, Y.; Zhang, L.; Yang, Y.; Liu, Y.; Yang, Z.; et al. APE1 deficiency promotes cellular senescence and premature aging features. Nucleic Acids Res. 2018, 46, 5664–5677. [Google Scholar] [CrossRef]
- Chen, D.; Cao, G.; Hastings, T.; Feng, Y.; Pei, W.; O’Horo, C.; Chen, J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J. Neurochem. 2002, 81, 1273–1284. [Google Scholar] [CrossRef]
- Chang, I.Y.; Lee, J.H.; Kim, J.N.; Lee, K.H.; Park, K.S.; Yoon, S.P. Apurinic/apyrimidinic endonuclease 1 on aging-associated deteriorations in rat kidneys. Free Radic. Res. 2015, 49, 95–101. [Google Scholar] [CrossRef]
- Kok, J.R.; Palminha, N.M.; Dos Santos Souza, C.; El-Khamisy, S.F.; Ferraiuolo, L. DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell. Mol. Life Sci. 2021, 78, 5707–5729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Du, X.; Sun, Z.; Xu, L. Expression of redox factor-1 in early injury period after liver transplantation in rat model. Cell. Mol. Immunol. 2009, 6, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Han, L.L.; Xie, L.P.; Li, L.H.; Zhang, X.W.; Zhang, R.Q.; Wang, H.Z. Reactive oxygen species production and Bax/Bcl-2 regulation in honokiol-induced apoptosis in human hepatocellular carcinoma SMMC-7721 cells. Env. Toxicol. Pharmacol. 2009, 28, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Raffoul, J.J.; Patel, H.V.; Prychitko, T.M.; Anyangwe, N.; Meira, L.B.; Friedberg, E.C.; Cabelof, D.C.; Heydari, A.R. Oxidative stress alters base excision repair pathway and increases apoptotic response in apurinic/apyrimidinic endonuclease 1/redox factor-1 haploinsufficient mice. Free Radic. Biol. Med. 2009, 46, 1488–1499. [Google Scholar] [CrossRef]
- Jiang, C.M.; Han, L.P.; Li, H.Z.; Qu, Y.B.; Zhang, Z.R.; Wang, R.; Xu, C.Q.; Li, W.M. Calcium-sensing receptors induce apoptosis in cultured neonatal rat ventricular cardiomyocytes during simulated ischemia/reperfusion. Cell Biol. Int. 2008, 32, 792–800. [Google Scholar] [CrossRef]
- Cursio, R.; Colosetti, P.; Auberger, P.; Gugenheim, J. Liver apoptosis following normothermic ischemia-reperfusion: In vivo evaluation of caspase activity by FLIVO assay in rats. Transpl. Proc. 2008, 40, 2038–2041. [Google Scholar] [CrossRef]
- Wang, B.Y.; Li, Q.X.; Li, J.; Xie, X.F.; Ao, Y.; Ai, Y.X. Hepatotoxicity and gene expression down-regulation of CYP isozymes caused by renal ischemia/reperfusion in the rat. Exp. Toxicol. Pathol. 2009, 61, 169–176. [Google Scholar] [CrossRef]
- Ozaki, M.; Suzuki, S.; Irani, K. Redox factor-1/APE suppresses oxidative stress by inhibiting the rac1 GTPase. FASEB J. 2002, 16, 889–890. [Google Scholar] [CrossRef]
- Sawakami, T.; Sun, Z.; Inagaki, Y.; Hasegawa, K.; Tang, W.; Xu, G.; Zhang, N. The impact of apurinic-apyrimidinic endonuclease I on hepatocyte immuno-inflammatory factors and cell apoptosis. Biosci. Trends 2019, 13, 539–545. [Google Scholar] [CrossRef]
- Ziolkowska, S.; Kosmalski, M.; Kolodziej, L.; Jablkowska, A.; Szemraj, J.Z.; Pietras, T.; Jablkowski, M.; Czarny, P.L. Single-Nucleotide Polymorphisms in Base-Excision Repair-Related Genes Involved in the Risk of an Occurrence of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2023, 24, 11307. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Dimayuga, P.C.; Zhao, X.; Nilsson, J.; Shah, P.K.; Cercek, B. Altered AP-1/Ref-1 redox pathway and reduced proliferative response in iNOS-deficient vascular smooth muscle cells. Vasc. Med. 2004, 9, 177–183. [Google Scholar] [CrossRef]
- Hou, X.; Snarski, P.; Higashi, Y.; Yoshida, T.; Jurkevich, A.; Delafontaine, P.; Sukhanov, S. Nuclear complex of glyceraldehyde-3-phosphate dehydrogenase and DNA repair enzyme apurinic/apyrimidinic endonuclease I protect smooth muscle cells against oxidant-induced cell death. FASEB J. 2017, 31, 3179–3192. [Google Scholar] [CrossRef] [PubMed]
- Kruta, M.; Balek, L.; Hejnova, R.; Dobsakova, Z.; Eiselleova, L.; Matulka, K.; Barta, T.; Fojtik, P.; Fajkus, J.; Hampl, A.; et al. Decrease in abundance of apurinic/apyrimidinic endonuclease causes failure of base excision repair in culture-adapted human embryonic stem cells. Stem Cells 2013, 31, 693–702. [Google Scholar] [CrossRef]
- Lee, K.M.; Lee, E.O.; Lee, Y.R.; Joo, H.K.; Park, M.S.; Kim, C.S.; Choi, S.; Jeong, J.O.; Jeon, B.H. APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2017, 18, 2053. [Google Scholar] [CrossRef] [PubMed]
- Bader, M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 439–465. [Google Scholar] [CrossRef]
- Juonala, M.; Viikari, J.S.; Ronnemaa, T.; Helenius, H.; Taittonen, L.; Raitakari, O.T. Elevated blood pressure in adolescent boys predicts endothelial dysfunction: The cardiovascular risk in young Finns study. Hypertension 2006, 48, 424–430. [Google Scholar] [CrossRef]
- Zheng, L.; Dai, Y.; Fu, P.; Yang, T.; Xie, Y.; Zheng, J.; Gao, J.; Niu, T. Secular trends of hypertension prevalence based on 2017 ACC/AHA and 2018 Chinese hypertension guidelines: Results from CHNS data (1991-2015). J. Clin. Hypertens. 2021, 23, 28–34. [Google Scholar] [CrossRef]
- Edelstein, L.C.; Lagos, L.; Simmons, M.; Tirumalai, H.; Gelinas, C. NF-kappa B-dependent assembly of an enhanceosome-like complex on the promoter region of apoptosis inhibitor Bfl-1/A1. Mol. Cell. Biol. 2003, 23, 2749–2761. [Google Scholar] [CrossRef] [PubMed]
- Sharp, K.J.; Sherar, L.B.; Kettle, V.E.; Sanders, J.P.; Daley, A.J. Effectiveness of interventions to increase device-measured physical activity in pregnant women: Systematic review and meta-analysis of randomised controlled trials. Int. J. Behav. Nutr. Phys. Act. 2022, 19, 142. [Google Scholar] [CrossRef] [PubMed]
- Lawes, C.M.; Vander Hoorn, S.; Rodgers, A. International Society of Hypertension. Global burden of blood-pressure-related disease, 2001. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Gong, Q.; Xie, J.; Li, Y.; Liu, Y.; Su, G. Enhanced ROBO4 is mediated by up-regulation of HIF-1alpha/SP1 or reduction in miR-125b-5p/miR-146a-5p in diabetic retinopathy. J. Cell. Mol. Med. 2019, 23, 4723–4737. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Dow, E.; Brennan, G.; Jung, R.T.; MacDonald, T.M. High prevalence of primary aldosteronism in the Tayside hypertension clinic population. J. Hum. Hypertens. 2000, 14, 311–315. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakayama, T.; Sato, N.; Fu, Z.; Soma, M.; Yamaguchi, M.; Shimodaira, M.; Aoi, N.; Usami, R. Haplotype-based case-control study on human apurinic/apyrimidinic endonuclease 1/redox effector factor-1 gene and essential hypertension. Am. J. Hypertens. 2010, 23, 186–191. [Google Scholar] [CrossRef]
- Jeon, B.H.; Gupta, G.; Park, Y.C.; Qi, B.; Haile, A.; Khanday, F.A.; Liu, Y.X.; Kim, J.M.; Ozaki, M.; White, A.R.; et al. Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ. Res. 2004, 95, 902–910. [Google Scholar] [CrossRef]
- Jung, S.B.; Kim, C.S.; Kim, Y.R.; Naqvi, A.; Yamamori, T.; Kumar, S.; Kumar, A.; Irani, K. Redox factor-1 activates endothelial SIRTUIN1 through reduction of conserved cysteine sulfhydryls in its deacetylase domain. PLoS ONE 2013, 8, e65415. [Google Scholar] [CrossRef]
- Lee, S.K.; Chung, J.I.; Park, M.S.; Joo, H.K.; Lee, E.J.; Cho, E.J.; Park, J.B.; Ryoo, S.; Irani, K.; Jeon, B.H. Apurinic/apyrimidinic endonuclease 1 inhibits protein kinase C-mediated p66shc phosphorylation and vasoconstriction. Cardiovasc. Res. 2011, 91, 502–509. [Google Scholar] [CrossRef]
- Sengupta, S.; Chattopadhyay, R.; Mantha, A.K.; Mitra, S.; Bhakat, K.K. Regulation of mouse-renin gene by apurinic/apyrimidinic-endonuclease 1 (APE1/Ref-1) via recruitment of histone deacetylase 1 corepressor complex. J. Hypertens. 2012, 30, 917–925. [Google Scholar] [CrossRef]
- Georgiadis, M.M.; Chen, Q.; Meng, J.; Guo, C.; Wireman, R.; Reed, A.; Vasko, M.R.; Kelley, M.R. Small molecule activation of apurinic/apyrimidinic endonuclease 1 reduces DNA damage induced by cisplatin in cultured sensory neurons. DNA. Repair 2016, 41, 32–41. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhang, Y.; Zhou, H.; Zhou, M.; Safford, M.M.; Muntner, P.; Moran, A.E.; Reynolds, K. Incidence of Atherosclerotic Cardiovascular Disease in Young Adults at Low Short-Term But High Long-Term Risk. J. Am. Coll. Cardiol. 2023, 81, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Simion, V.; Zhou, H.; Haemmig, S.; Pierce, J.B.; Mendes, S.; Tesmenitsky, Y.; Perez-Cremades, D.; Lee, J.F.; Chen, A.F.; Ronda, N.; et al. A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nat. Commun. 2020, 11, 6135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, J.; Wang, M.; Zhang, X.; Zhou, M. Epidemiology of cardiovascular disease in China: Current features and implications. Nat. Rev. Cardiol. 2019, 16, 203–212. [Google Scholar] [CrossRef]
- Dos Santos, L.; Bertoli, S.R.; Avila, R.A.; Marques, V.B. Iron overload, oxidative stress and vascular dysfunction: Evidences from clinical studies and animal models. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130172. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Godschalk, R.W.; Albrecht, C.; Curfs, D.M.; Schins, R.P.; Bartsch, H.; van Schooten, F.J.; Nair, J. Decreased levels of lipid peroxidation-induced DNA damage in the onset of atherogenesis in apolipoprotein E deficient mice. Mutat. Res. 2007, 621, 87–94. [Google Scholar] [CrossRef]
- Dai, J.; Li, W.; Chang, L.; Zhang, Z.; Tang, C.; Wang, N.; Zhu, Y.; Wang, X. Role of redox factor-1 in hyperhomocysteinemia-accelerated atherosclerosis. Free. Radic. Biol. Med. 2006, 41, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Joo, H.K.; Lee, E.O.; Park, M.S.; Cho, H.S.; Kim, S.; Jin, H.; Jeong, J.O.; Kim, C.S.; Jeon, B.H. Plasma APE1/Ref-1 Correlates with Atherosclerotic Inflammation in ApoE(-/-) Mice. Biomedicines 2020, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Son, S.J.; Kim, E.K.; Kim, S.N.; Yoo, D.G.; Kim, H.S.; Ryoo, S.W.; Lee, S.D.; Irani, K.; Jeon, B.H. Apurinic/apyrimidinic endonuclease1/redox factor-1 inhibits monocyte adhesion in endothelial cells. Cardiovasc. Res. 2006, 69, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, A.; Kawabe, J.; Kabara, M.; Matsuki, M.; Asanome, A.; Aonuma, T.; Ohta, H.; Takehara, N.; Kitagawa, T.; Hasebe, N. Apurinic/apyrimidinic endonucelase 1 maintains adhesion of endothelial progenitor cells and reduces neointima formation. Am. J. Physiol. Heart. Circ. Physiol. 2013, 305, H1158–H1167. [Google Scholar] [CrossRef]
- Merk, D.; Ptok, J.; Jakobs, P.; von Ameln, F.; Greulich, J.; Kluge, P.; Semperowitsch, K.; Eckermann, O.; Schaal, H.; Ale-Agha, N.; et al. Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis. Antioxidants 2021, 10, 1472. [Google Scholar] [CrossRef]
- Xu, H.; Tan, L.; Qu, Q.; Zhang, W. NEDD4 attenuates oxidized low-density lipoprotein-induced inflammation and dysfunction in vascular endothelial cells via regulating APEX1 expression. Exp. Ther. Med. 2023, 25, 88. [Google Scholar] [CrossRef]
- Joo, H.K.; Lee, Y.R.; Park, M.S.; Choi, S.; Park, K.; Lee, S.K.; Kim, C.S.; Park, J.B.; Jeon, B.H. Mitochondrial APE1/Ref-1 suppressed protein kinase C-induced mitochondrial dysfunction in mouse endothelial cells. Mitochondrion 2014, 17, 42–49. [Google Scholar] [CrossRef]
- Luo, M.; He, H.; Kelley, M.R.; Georgiadis, M.M. Redox regulation of DNA repair: Implications for human health and cancer therapeutic development. Antioxid. Redox Signal 2010, 12, 1247–1269. [Google Scholar] [CrossRef]
- Zhao, C.R.; Yang, F.F.; Cui, Q.; Wang, D.; Zhou, Y.; Li, Y.S.; Zhang, Y.P.; Tang, R.Z.; Yao, W.J.; Wang, X.; et al. Vitexin inhibits APEX1 to counteract the flow-induced endothelial inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2115158118. [Google Scholar] [CrossRef]
- Bautista-Nino, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.; Roks, A.J. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakayama, T.; Sato, N.; Fu, Z.; Yamaguchi, M.; Soma, M.; Aoi, N.; Usami, R.; Doba, N.; Hinohara, S. Haplotype-based case-control study between human apurinic/apyrimidinic endonuclease 1/redox effector factor-1 gene and cerebral infarction. Clin. Biochem. 2009, 42, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Venkat, P.; Chopp, M.; Zacharek, A.; Yu, P.; Ning, R.; Qiao, X.; Kelley, M.R.; Chen, J. APX3330 Promotes Neurorestorative Effects after Stroke in Type One Diabetic Rats. Aging. Dis. 2018, 9, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Englander, E.W. Nuclear depletion of apurinic/apyrimidinic endonuclease 1 (Ape1/Ref-1) is an indicator of energy disruption in neurons. Free. Radic. Biol. Med. 2012, 53, 1782–1790. [Google Scholar] [CrossRef]
- Yang, J.L.; Chen, W.Y.; Chen, Y.P.; Kuo, C.Y.; Chen, S.D. Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1. Theranostics 2016, 6, 2015–2027. [Google Scholar] [CrossRef]
- Stetler, R.A.; Gao, Y.; Leak, R.K.; Weng, Z.; Shi, Y.; Zhang, L.; Pu, H.; Zhang, F.; Hu, X.; Hassan, S.; et al. APE1/Ref-1 facilitates recovery of gray and white matter and neurological function after mild stroke injury. Proc. Natl. Acad. Sci. USA 2016, 113, E3558–E3567. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Kim, J.E.; Park, W.H. Cytoprotective effect of rhamnetin on miconazole-induced H9c2 cell damage. Nutr. Res. Pr. 2015, 9, 586–591. [Google Scholar] [CrossRef]
- Li, X.; Le, H.T.; Sato, F.; Kang, T.H.; Makishima, M.; Zhong, L.; Liu, Y.; Guo, L.; Bhawal, U.K. Dec1 deficiency protects the heart from fibrosis, inflammation, and myocardial cell apoptosis in a mouse model of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2020, 532, 513–519. [Google Scholar] [CrossRef]
- Won, K.J.; Lin, H.Y.; Jung, S.; Cho, S.M.; Shin, H.C.; Bae, Y.M.; Lee, S.H.; Kim, H.J.; Jeon, B.H.; Kim, B. Antifungal miconazole induces cardiotoxicity via inhibition of APE/Ref-1-related pathway in rat neonatal cardiomyocytes. Toxicol. Sci. 2012, 126, 298–305. [Google Scholar] [CrossRef]
- Tang, W.; Lin, D.; Chen, M.; Li, Z.; Zhang, W.; Hu, W.; Li, F. PTEN-mediated mitophagy and APE1 overexpression protects against cardiac hypoxia/reoxygenation injury. Vitr. Cell. Dev. Biol. Anim. 2019, 55, 741–748. [Google Scholar] [CrossRef]
- Uddin, M.A.; Akhter, M.S.; Siejka, A.; Catravas, J.D.; Barabutis, N. P53 supports endothelial barrier function via APE1/Ref1 suppression. Immunobiology 2019, 224, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Dyballa-Rukes, N.; Jakobs, P.; Eckers, A.; Ale-Agha, N.; Serbulea, V.; Aufenvenne, K.; Zschauer, T.C.; Rabanter, L.L.; Jakob, S.; von Ameln, F.; et al. The Anti-Apoptotic Properties of APEX1 in the Endothelium Require the First 20 Amino Acids and Converge on Thioredoxin-1. Antioxid. Redox Signal 2017, 26, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Won, K.J.; Lee, K.P.; Jung, S.H.; Baek, S.; Chung, H.W.; Choi, W.S.; Lee, H.M.; Lee, B.H.; Jeon, B.H.; et al. Angiotensin II facilitates neointimal formation by increasing vascular smooth muscle cell migration: Involvement of APE/Ref-1-mediated overexpression of sphingosine-1-phosphate receptor 1. Toxicol. Appl. Pharmacol. 2018, 347, 45–53. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Weintraub, N.L.; Goswami, P.C.; Chatterjee, P.; Flaherty, D.M.; Domann, F.E.; Oberley, L.W. Redox factor-1 contributes to the regulation of progression from G0/G1 to S by PDGF in vascular smooth muscle cells. Am. J. Physiol. Heart. Circ. Physiol. 2003, 285, H804–H812. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal 2009, 11, 621–638. [Google Scholar] [CrossRef]
- London, G.M.; Guerin, A.P.; Marchais, S.J.; Metivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transpl. 2003, 18, 1731–1740. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.V.; Mokas, S.; Lariviere, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef]
- Torres-Gonzalez, M.; Gawlowski, T.; Kocalis, H.; Scott, B.T.; Dillmann, W.H. Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in H9C2 cells under oxidative stress conditions. Am. J. Physiol. Cell. Physiol. 2014, 306, C221–C229. [Google Scholar] [CrossRef]
- Zhang, J.; Geng, Y.; Guo, F.; Zhang, F.; Liu, M.; Song, L.; Ma, Y.; Li, D.; Zhang, Y.; Xu, H.; et al. Screening and identification of critical transcription factors involved in the protection of cardiomyocytes against hydrogen peroxide-induced damage by Yixin-shu. Sci. Rep. 2017, 7, 13867. [Google Scholar] [CrossRef]
- Fan, J.; Liu, M.; Li, X.; Gao, S.; Wang, Y.; Li, A.; Chen, L.; Zhou, D.; Chen, H.; Xu, Z.; et al. Apurinic/apyrimidinic endonuclease 1 regulates palmitic acid-mediated apoptosis in cardiomyocytes via endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2023, 650, 123–131. [Google Scholar] [CrossRef]
- Gurusamy, N.; Malik, G.; Gorbunov, N.V.; Das, D.K. Redox activation of Ref-1 potentiates cell survival following myocardial ischemia reperfusion injury. Free. Radic. Biol. Med. 2007, 43, 397–407. [Google Scholar] [CrossRef]
- Yamagami, I.; Suzuki, Y.; Ito, K. Pharmacological studies on the components of Fraxinus japonica Blume. Nihon. Yakurigaku Zasshi 1968, 64, 714–729. [Google Scholar] [PubMed]
- Jin, S.A.; Lim, B.K.; Seo, H.J.; Kim, S.K.; Ahn, K.T.; Jeon, B.H.; Jeong, J.O. Elevation of Serum APE1/Ref-1 in Experimental Murine Myocarditis. Int. J. Mol. Sci. 2017, 18, 2664. [Google Scholar] [CrossRef] [PubMed]
- Tekeli, A.; Isbir, S.; Ergen, A.; Gormus, U.; Bozkurt, N.; Timirci, O.; Arsan, S.; Isbir, T. APE1 and XRCC3 polymorphisms and myocardial infarction. In Vivo 2008, 22, 477–479. [Google Scholar] [PubMed]
- Hu, J.J.; Smith, T.R.; Miller, M.S.; Mohrenweiser, H.W.; Golden, A.; Case, L.D. Amino acid substitution variants of APE1 and XRCC1 genes associated with ionizing radiation sensitivity. Carcinogenesis 2001, 22, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Aonuma, T.; Takehara, N.; Maruyama, K.; Kabara, M.; Matsuki, M.; Yamauchi, A.; Kawabe, J.; Hasebe, N. Apoptosis-Resistant Cardiac Progenitor Cells Modified With Apurinic/Apyrimidinic Endonuclease/Redox Factor 1 Gene Overexpression Regulate Cardiac Repair After Myocardial Infarction. Stem. Cells. Transl. Med. 2016, 5, 1067–1078. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, H.-H.; Yin, H.; Marincas, M.; Xie, L.-L.; Bu, L.-L.; Guo, M.-H.; Zheng, X.-L. From DNA Repair to Redox Signaling: The Multifaceted Role of APEX1 (Apurinic/Apyrimidinic Endonuclease 1) in Cardiovascular Health and Disease. Int. J. Mol. Sci. 2025, 26, 3034. https://doi.org/10.3390/ijms26073034
Yuan H-H, Yin H, Marincas M, Xie L-L, Bu L-L, Guo M-H, Zheng X-L. From DNA Repair to Redox Signaling: The Multifaceted Role of APEX1 (Apurinic/Apyrimidinic Endonuclease 1) in Cardiovascular Health and Disease. International Journal of Molecular Sciences. 2025; 26(7):3034. https://doi.org/10.3390/ijms26073034
Chicago/Turabian StyleYuan, Huan-Huan, Hao Yin, Mara Marincas, Ling-Li Xie, Lan-Lan Bu, Min-Hua Guo, and Xi-Long Zheng. 2025. "From DNA Repair to Redox Signaling: The Multifaceted Role of APEX1 (Apurinic/Apyrimidinic Endonuclease 1) in Cardiovascular Health and Disease" International Journal of Molecular Sciences 26, no. 7: 3034. https://doi.org/10.3390/ijms26073034
APA StyleYuan, H.-H., Yin, H., Marincas, M., Xie, L.-L., Bu, L.-L., Guo, M.-H., & Zheng, X.-L. (2025). From DNA Repair to Redox Signaling: The Multifaceted Role of APEX1 (Apurinic/Apyrimidinic Endonuclease 1) in Cardiovascular Health and Disease. International Journal of Molecular Sciences, 26(7), 3034. https://doi.org/10.3390/ijms26073034