
Molecular Basis of High-Blood-Pressure-Enhanced and High-Fever-Temperature-Weakened Receptor-Binding Domain/Peptidase Domain Binding: A Molecular Dynamics Simulation Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
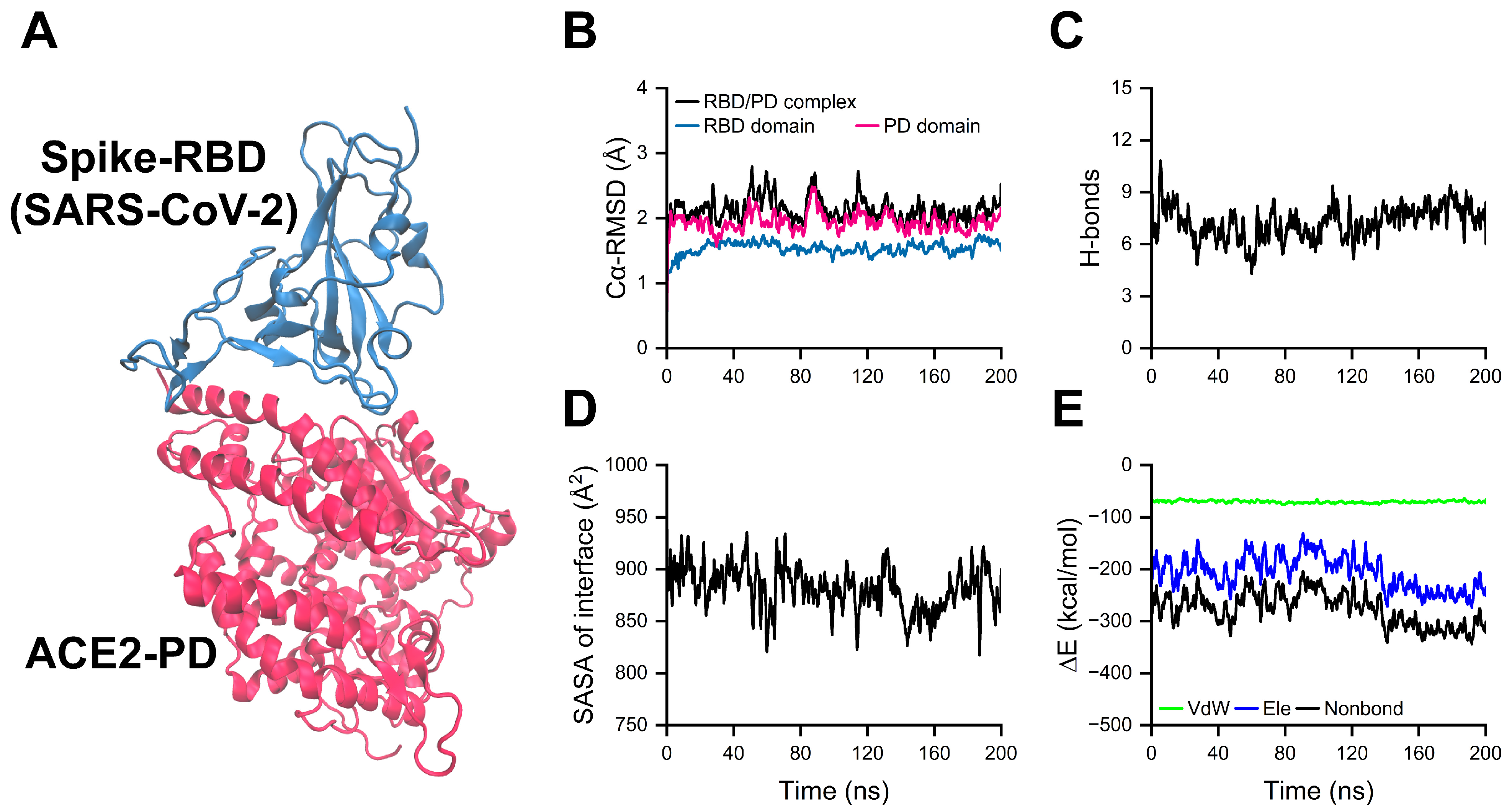
2.1. Equilibration of RBD/PD Complexes
2.2. High Pressure Enhances RBD/PD Binding
2.3. Curving of α1-Helix in the PD Mediates High-Pressure-Enhanced RBD/PD Binding
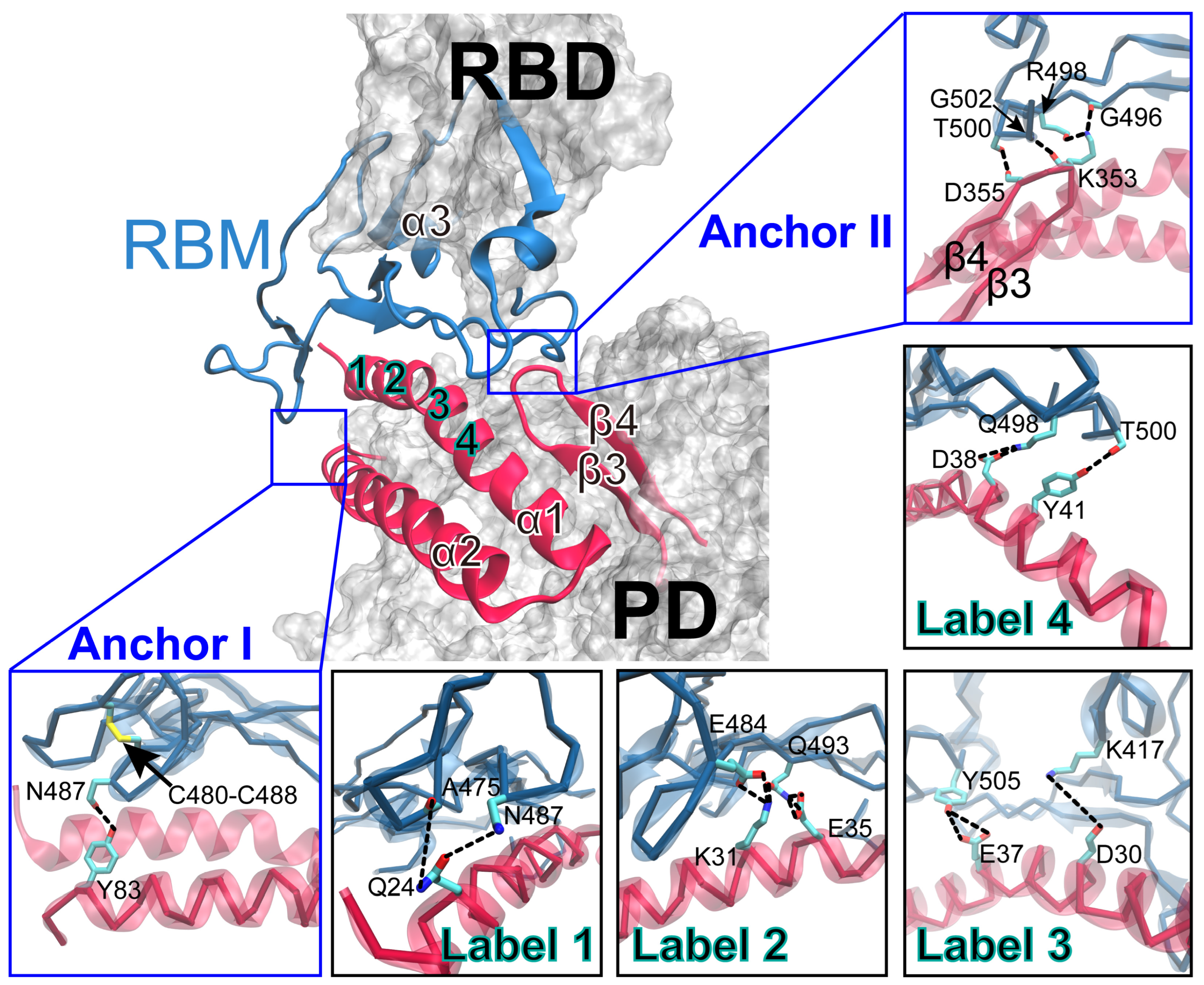
2.4. Key Residues Underlying High-Pressure-Upregulated RBD/PD Binding
2.5. High Temperature Weakens and Low Temperature Enhances RBD/PD Binding
2.6. Straightening of the α1-Helix in the PD Mediates High-Temperature Weakening of RBD/PD Binding
2.7. Key Residues Underlying High-Temperature-Downregulated RBD/PD Binding
3. Discussion
4. Materials and Methods
4.1. System Setup
4.2. Minimization and Equilibrium of Complex Systems
4.3. Pressure and Temperature Simulations
4.4. Data Analyses
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.; Wang, W.; Song, Z.; Hu, Y.; Tao, Z.; Tian, J.; Pei, Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Steiner, S.; Kratzel, A.; Barut, G.T.; Lang, R.M.; Aguiar Moreira, E.; Thomann, L.; Kelly, J.N.; Thiel, V. SARS-CoV-2 biology and host interactions. Nat. Rev. Microbiol. 2024, 22, 206–225. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin converting enzyme 2 a double-edged sword. Circulation 2020, 142, 426–428. [Google Scholar] [CrossRef]
- Li, W.H.; Moore, M.J.; Vasilieva, N.; Sui, J.H.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Wang, K.; Viveiros, A.; Kellner, M.J.; Penninger, J.M. Angiotensin-converting enzyme 2-at the heart of the COVID-19 pandemic. Cell 2023, 186, 906–922. [Google Scholar] [CrossRef]
- NCD-RisC, N.R.F.C. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef]
- Ravichandran, B.; Grimm, D.; Krüger, M.; Kopp, S.; Infanger, M.; Wehland, M. SARS-CoV-2 and hypertension. Physiol. Rep. 2021, 9, e14800. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C.; Grassi, G.; Mancia, G. COVID-19 and arterial hypertension: Hypothesis or evidence? J. Clin. Hypertens. 2020, 22, 1120–1126. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Kompaniyets, L.; Pennington, A.F.; Goodman, A.B.; Rosenblum, H.G.; Belay, B.; Ko, J.Y.; Chevinsky, J.R.; Schieber, L.Z.; Summers, A.D.; Lavery, A.M.; et al. Underlying medical conditions and severe illness among 540,667 adults hospitalized with COVID-19, March 2020-March 2021. Prev. Chronic Dis. 2021, 18, E66. [Google Scholar] [CrossRef]
- Huang, S.; Wang, J.; Liu, F.; Liu, J.; Cao, G.; Yang, C.; Liu, W.; Tu, C.; Zhu, M.; Xiong, B. COVID-19 patients with hypertension have more severe disease: A multicenter retrospective observational study. Hypertens. Res. 2020, 43, 824–831. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, J.; Sun, X.; Xue, H.; Shao, J.; Cai, W.; Jing, Y.; Yue, M.; Dong, C. Association of hypertension with the severity and fatality of SARS-CoV-2 infection: A meta-analysis. Epidemiol. Infect. 2020, 148, e106. [Google Scholar] [CrossRef]
- Savoia, C.; Volpe, M.; Kreutz, R. Hypertension, a moving target in COVID-19: Current views and perspectives. Circ. Res. 2021, 128, 1062–1079. [Google Scholar] [CrossRef]
- Dessie, Z.G.; Zewotir, T. Mortality-related risk factors of COVID-19: A systematic review and meta-analysis of 42 studies and 423,117 patients. BMC Infect. Dis. 2021, 21, 855. [Google Scholar] [CrossRef]
- Yan, L.D.; Matuja, S.S.; Pain, K.J.; McNairy, M.L.; Etyang, A.O.; Peck, R.N. Emerging viral infections, hypertension, and cardiovascular disease in Sub-Saharan Africa: A narrative review. Hypertension 2022, 79, 898–905. [Google Scholar] [CrossRef]
- Kabia, A.U.; Li, P.; Jin, Z.; Tan, X.; Liu, Y.; Feng, Y.; Yu, K.; Hu, M.; Jiang, D.; Cao, G. The effects of hypertension on the prognosis of coronavirus disease 2019: A systematic review and meta-analysis on the interactions with age and antihypertensive treatment. J. Hypertens. 2022, 40, 2323–2336. [Google Scholar] [CrossRef]
- D’Elia, L.; Giaquinto, A.; Zarrella, A.F.; Rendina, D.; Iaccarino, I.P.; Strazzullo, P.; Galletti, F. Hypertension and mortality in SARS-COV-2 infection: A meta-analysis of observational studies after 2 years of pandemic. Eur. J. Intern. Med. 2023, 108, 28–36. [Google Scholar] [CrossRef]
- Schiffrin, E.L. How Structure, Mechanics, and Function of the Vasculature Contribute to Blood Pressure Elevation in Hypertension. Can. J. Cardiol. 2020, 36, 648–658. [Google Scholar] [CrossRef]
- Boutouyrie, P.; Chowienczyk, P.; Humphrey, J.D.; Mitchell, G.F. Arterial Stiffness and Cardiovascular Risk in Hypertension. Circ. Res. 2021, 128, 864–886. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Fei, P.; Zhang, T.; Yao, D.; Gao, Y.; Liu, J.; Chen, H.; Lu, Q.; Mudianto, T.; et al. Mechanical activation of spike fosters SARS-CoV-2 viral infection. Cell Res. 2021, 31, 1047–1060. [Google Scholar] [CrossRef]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef]
- Yu, C.; Lei, Q.; Li, W.; Wang, X.; Liu, W.; Fan, X.; Li, W. Clinical characteristics, associated factors, and predicting COVID-19 mortality risk: A retrospective study in Wuhan, China. Am. J. Prev. Med. 2020, 59, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Tu, W.J.; Cheng, W.; Yu, L.; Liu, Y.K.; Hu, X.; Liu, Q. Clinical features and short-term outcomes of 102 patients with coronavirus disease 2019 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Fatteh, N.; Sutherland, G.E.; Santos, R.G.; Zeidan, R.; Gastesi, A.P.; Naranjo, C.D. Association of hypothermia with increased mortality rate in SARS-CoV-2 infection. Int. J. Infect. Dis. 2021, 108, 167–170. [Google Scholar] [CrossRef]
- Tofan, V.; Lenghel, A.; de Camargo, M.M.; Stan, R.C. Fever as an evolutionary agent to select immune complexes interfaces. Immunogenetics 2022, 74, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Tharakan, S.; Nomoto, K.; Miyashita, S.; Ishikawa, K. Body temperature correlates with mortality in COVID-19 patients. Crit. Care 2020, 24, 298. [Google Scholar] [CrossRef]
- Kim, D.G.; Kim, H.S.; Choi, Y.; Stan, R.C. Fever temperatures modulate intraprotein dynamics and enhance the binding affinity between monoclonal antibodies and the spike protein from SARS-CoV-2. Comp. Struct. Biotechnol. J. 2022, 20, 5962–5965. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, Z.; Ou, J.; Zhang, H.; Zhang, Q.; Dong, M.; Zhang, G. Temperature dependence of the SARS-CoV-2 affinity to human ACE2 determines COVID-19 progression and clinical outcome. Comp. Struct. Biotechnol. J. 2021, 19, 161–167. [Google Scholar] [CrossRef]
- Drewry, A.M.; Hotchkiss, R.; Kulstad, E. Response to “Body temperature correlates with mortality in COVID-19 patients”. Crit. Care 2020, 24, 460. [Google Scholar] [CrossRef]
- Benlarbi, M.; Ding, S.; Belanger, E.; Tauzin, A.; Poujol, R.; Medjahed, H.; El, F.O.; Bo, Y.; Bourassa, C.; Hussin, J.; et al. Temperature-dependent Spike-ACE2 interaction of Omicron subvariants is associated with viral transmission. mBio 2024, 15, e0090724. [Google Scholar] [CrossRef]
- Prevost, J.; Richard, J.; Gasser, R.; Ding, S.; Fage, C.; Anand, S.P.; Adam, D.; Gupta, V.N.; Tauzin, A.; Benlarbi, M.; et al. Impact of temperature on the affinity of SARS-CoV-2 Spike glycoprotein for host ACE2. J. Biol. Chem. 2021, 297, 101151. [Google Scholar] [CrossRef]
- Gong, S.Y.; Ding, S.; Benlarbi, M.; Chen, Y.; Vezina, D.; Marchitto, L.; Beaudoin-Bussieres, G.; Goyette, G.; Bourassa, C.; Bo, Y.; et al. Temperature influences the interaction between SARS-CoV-2 spike from Omicron subvariants and human ACE2. Viruses 2022, 14, 2178. [Google Scholar] [CrossRef]
- Forest-Nault, C.; Koyuturk, I.; Gaudreault, J.; Pelletier, A.; L Abbé, D.; Cass, B.; Bisson, L.; Burlacu, A.; Delafosse, L.; Stuible, M.; et al. Impact of the temperature on the interactions between common variants of the SARS-CoV-2 receptor binding domain and the human ACE2. Sci. Rep. 2022, 12, 11520. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, Z.; Fang, Y.; Wu, J. Prediction of catch-slip bond transition of Kindlin2/β3 integrin via steered molecular dynamics simulation. J. Chem. Inf. Model. 2020, 60, 5132–5141. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Wu, J.; Fang, Y. Moderate constraint facilitates association and force-dependent dissociation of HA-CD44 complex. Int. J. Mol. Sci. 2023, 24, 2243. [Google Scholar] [CrossRef]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long covid-mechanisms, risk factors, and management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef]
- Ashraf, U.M.; Abokor, A.A.; Edwards, J.M.; Waigi, E.W.; Royfman, R.S.; Hasan, S.A.; Smedlund, K.B.; Hardy, A.; Chakravarti, R.; Koch, L.G. SARS-CoV-2, ACE2 expression, and systemic organ invasion. Physiol. Genom. 2021, 53, 51–60. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Li, W.; Xu, Z.; Niu, T.; Xie, Y.; Zhao, Z.; Li, D.; He, Q.; Sun, W.; Shi, K.; Guo, W.; et al. Key mechanistic features of the trade-off between antibody escape and host cell binding in the SARS-CoV-2 Omicron variant spike proteins. EMBO J. 2024, 43, 1484–1498. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y.; et al. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, K.; Gu, Y.; Xu, Z.; Shu, C.; Li, D.; Sun, J.; Cong, M.; Li, X.; Zhao, X.; et al. Spike structures, receptor binding, and immune escape of recently circulating SARS-CoV-2 Omicron BA.2.86, JN.1, EG.5, EG.5.1, and HV.1 sub-variants. Structure 2024, 32, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Chen, X.; Xu, Y.; Wang, P.; Wang, J.; Yu, L.; et al. Antigenicity and infectivity characterisation of SARS-CoV-2 BA.2.86. Lancet Infect. Dis. 2023, 23, e457–e459. [Google Scholar] [CrossRef]
- Uriu, K.; Ito, J.; Kosugi, Y.; Tanaka, Y.L.; Mugita, Y.; Guo, Z.; Hinay, A.A.; Putri, O.; Kim, Y.; Shimizu, R.; et al. Transmissibility, infectivity, and immune evasion of the SARS-CoV-2 BA.2.86 variant. Lancet Infect. Dis. 2023, 23, e460–e461. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Y.; Xu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Wang, P.; Wang, J.; Liu, J.; Yu, L.; et al. Fast evolution of SARS-CoV-2 BA.2.86 to JN.1 under heavy immune pressure. Lancet Infect. Dis. 2024, 24, e70–e72. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, D.; Dijokaite-Guraliuc, A.; Supasa, P.; Duyvesteyn, H.; Ginn, H.M.; Selvaraj, M.; Mentzer, A.J.; Das, R.; de Silva, T.I.; et al. A structure-function analysis shows SARS-CoV-2 BA.2.86 balances antibody escape and ACE2 affinity. Cell Rep. Med. 2024, 5, 101553. [Google Scholar] [CrossRef]
- Jian, F.; Wang, J.; Yisimayi, A.; Song, W.; Xu, Y.; Chen, X.; Niu, X.; Yang, S.; Yu, Y.; Wang, P.; et al. Evolving antibody response to SARS-CoV-2 antigenic shift from XBB to JN.1. Nature 2025, 637, 921–929. [Google Scholar] [CrossRef]
- Rath, S.L.; Kumar, K. Investigation of the Effect of Temperature on the Structure of SARS-CoV-2 Spike Protein by Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 7, 583523. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. Elife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Turonova, B.; Sikora, M.; Schurmann, C.; Hagen, W.; Welsch, S.; Blanc, F.; von Bulow, S.; Gecht, M.; Bagola, K.; Horner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 44130. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.J. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Vidal-Petiot, E. Thresholds for hypertension definition, treatment initiation, and treatment targets: Recent guidelines at a glance. Circulation 2022, 146, 805–807. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Sarkar, S.; Zhang, J.; Witham, S.; Zhang, Z.; Wang, L.; Smith, N.; Petukh, M.; Alexov, E. DelPhi: A comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012, 5, 9. [Google Scholar] [CrossRef]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2021, 22, bbaa161. [Google Scholar] [CrossRef]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, X.; Zhang, Y.; Fang, Y.; Wu, J.; Li, Q. Molecular Basis of High-Blood-Pressure-Enhanced and High-Fever-Temperature-Weakened Receptor-Binding Domain/Peptidase Domain Binding: A Molecular Dynamics Simulation Study. Int. J. Mol. Sci. 2025, 26, 3250. https://doi.org/10.3390/ijms26073250
Xie X, Zhang Y, Fang Y, Wu J, Li Q. Molecular Basis of High-Blood-Pressure-Enhanced and High-Fever-Temperature-Weakened Receptor-Binding Domain/Peptidase Domain Binding: A Molecular Dynamics Simulation Study. International Journal of Molecular Sciences. 2025; 26(7):3250. https://doi.org/10.3390/ijms26073250
Chicago/Turabian StyleXie, Xubin, Yu Zhang, Ying Fang, Jianhua Wu, and Quhuan Li. 2025. "Molecular Basis of High-Blood-Pressure-Enhanced and High-Fever-Temperature-Weakened Receptor-Binding Domain/Peptidase Domain Binding: A Molecular Dynamics Simulation Study" International Journal of Molecular Sciences 26, no. 7: 3250. https://doi.org/10.3390/ijms26073250
APA StyleXie, X., Zhang, Y., Fang, Y., Wu, J., & Li, Q. (2025). Molecular Basis of High-Blood-Pressure-Enhanced and High-Fever-Temperature-Weakened Receptor-Binding Domain/Peptidase Domain Binding: A Molecular Dynamics Simulation Study. International Journal of Molecular Sciences, 26(7), 3250. https://doi.org/10.3390/ijms26073250