
In Silico Identification of Putative Allosteric Pockets and Inhibitors for the KRASG13D-SOS1 Complex in Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
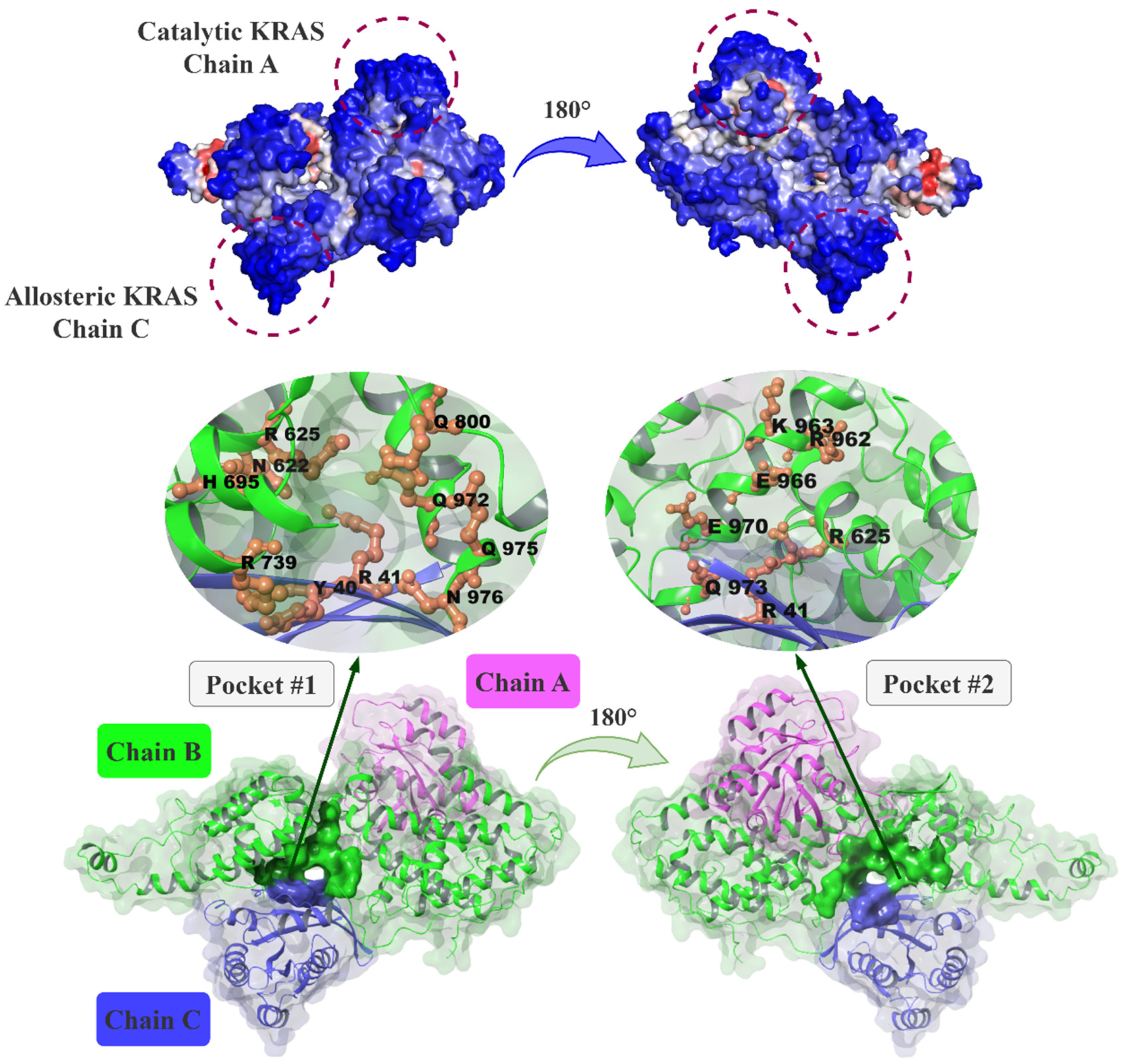
2.1. Prediction of Putative Allosteric Binding Sites on SOS1 and Its Complexes
2.2. Molecular Docking Calculations for the Known Binding Site and Two Putative Allosteric Binding Sites
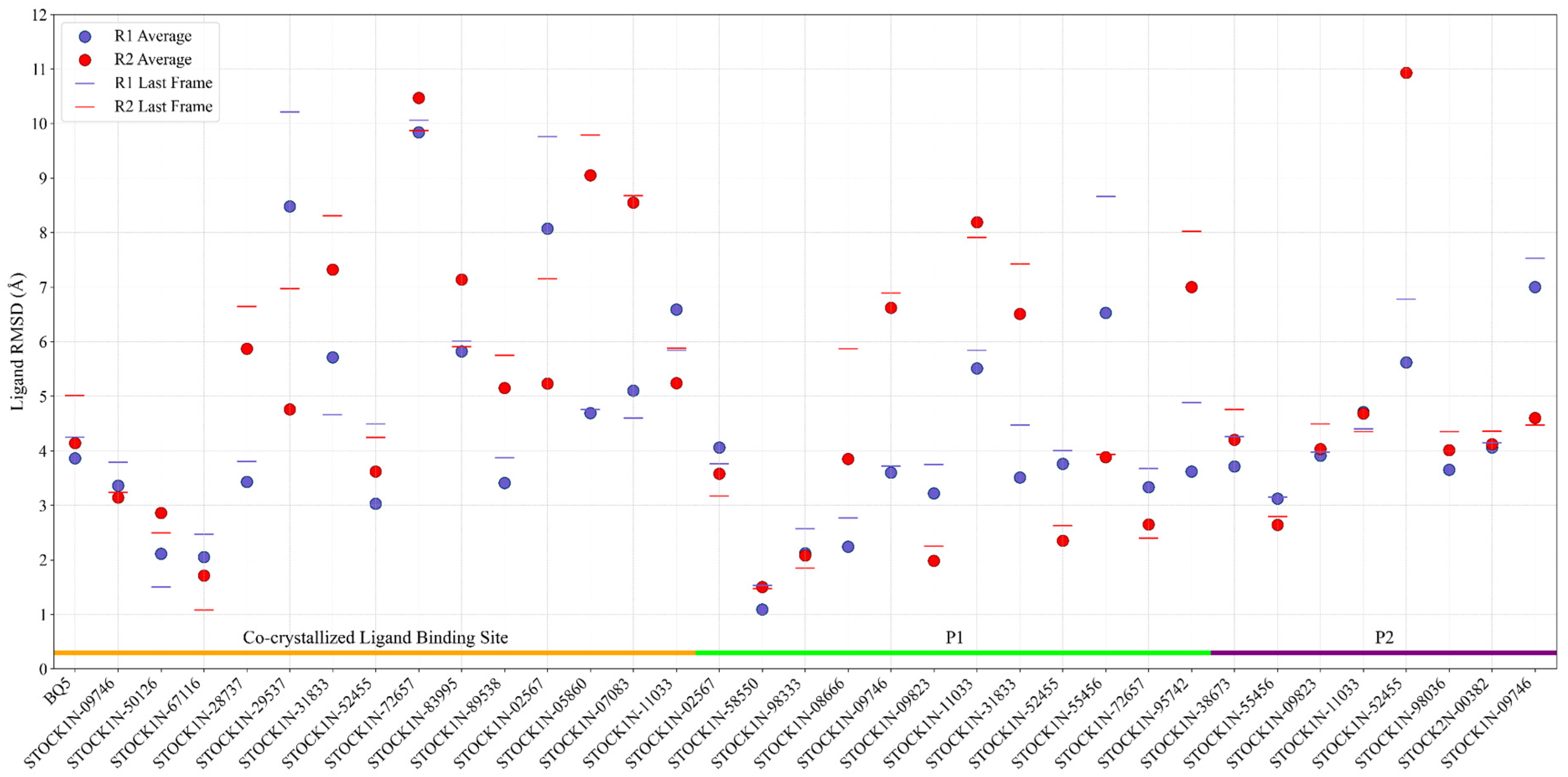
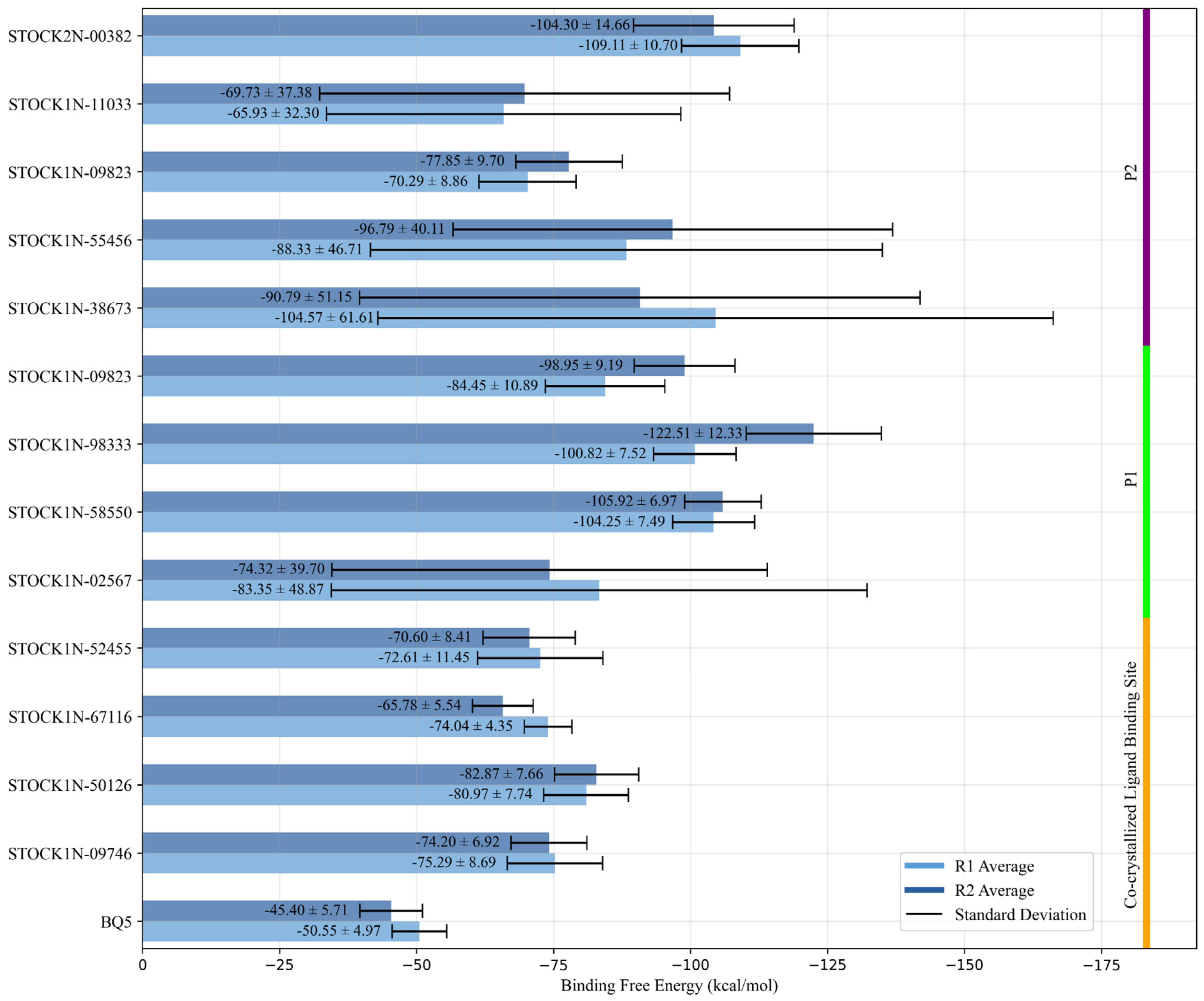
2.3. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Essential Site Scanning Analysis
3.2. Residue Interaction Network Model
3.3. Dataset Preparation
3.4. Structure-Based Virtual Screening with Glide
3.5. Prime MM-GBSA Calculations in Glide
3.6. Molecular Dynamics Simulations
3.7. Prediction of the Pharmacokinetic Properties and Toxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrem, J.M.L.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Orgován, Z.; Keserű, G.M. Small molecule inhibitors of RAS proteins with oncogenic mutations. Cancer Metastasis Rev. 2020, 39, 1107–1126. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Wang, Y.; Chai, Z.; Ni, D.; Li, X.; Pu, J.; Chen, J.; Zhang, J.; Lu, S.; Lv, C.; et al. Targeting RAS phosphorylation in cancer therapy: Mechanisms and modulators. Acta Pharm. Sin. B 2021, 11, 3433–3446. [Google Scholar] [CrossRef] [PubMed]
- Mattox, T.E.; Chen, X.; Maxuitenko, Y.Y.; Keeton, A.B.; Piazza, G.A. Exploiting RAS nucleotide cycling as a strategy for drugging RAS-driven cancers. Int. J. Mol. Sci. 2019, 21, 141. [Google Scholar] [CrossRef]
- Roman, M.; Hwang, E.; Sweet-Cordero, E.A. Synthetic vulnerabilities in the KRAS pathway. Cancers 2022, 14, 2837. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.W.; Lin, Y.J.; Reid, D.; Parker, J.; Pavlopoulos, S.; Dischinger, P.; Graveel, C.; Aguirre, A.J.; Steensma, M.; Haigis, K.M.; et al. Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell Rep. 2019, 28, 1538–1550.e7. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.; Shannon, K.M.; Bollag, G. GTPase activating proteins: Critical regulators of intracellular signaling. Biochim. Biophys. Acta Rev. Cancer 2002, 1602, 23–45. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Gu, S.; Zhang, J.; Nussinov, R. Drugging Ras GTPase: A comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 2016, 45, 4929–4952. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Morrison, D.K. A Structure Is Worth a Thousand Words: New Insights for RAS and RAF Regulation. Cancer Discov. 2022, 12, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Banerjee, A.; Jang, H.; Zhang, J.; Gaponenko, V.; Nussinov, R. GTP binding and oncogenic mutations may attenuate hypervariable region (HVR)-catalytic domain interactions in small GTPase K-Ras4B, exposing the effector binding site. J. Biol. Chem. 2015, 290, 28887–28900. [Google Scholar] [CrossRef]
- Eren, M.; Tuncbag, N.; Jang, H.; Nussinov, R.; Gursoy, A.; Keskin, O. Normal Mode Analysis of KRas4B Reveals Partner Specific Dynamics. J. Phys. Chem. B 2021, 125, 5210–5221. [Google Scholar] [CrossRef] [PubMed]
- Trahey, M.; McCormick, F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987, 238, 542–545. [Google Scholar]
- Xu, G.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [PubMed]
- Ahmadian, M.R.; Hoffmann, U.; Goody, R.S.; Wittinghofer, A. Individual rate constants for the interaction of Ras proteins with GTPase-activating proteins determined by fluorescence spectroscopy. Biochemistry 1997, 36, 4535–4541. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Vaghi, C.; Baraibar, I.; Saoudi González, N.; Rodríguez-Castells, M.; García, A.; Alcaraz, A.; Salva, F.; Tabernero, J.; Elez, E. Targeting KRAS G12C Mutation in Colorectal Cancer, A Review: New Arrows in the Quiver. Int. J. Mol. Sci. 2024, 25, 3304. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.M.; Tu, H.-L.; Jun, J.E.; Alvarez, S.; Triplet, M.G.; Iwig, J.S.; Yadav, K.K.; Bar-Sagi, D.; Roose, J.P.; Groves, J.T. One-way membrane trafficking of SOS in receptor-triggered Ras activation. Nat. Struct. Mol. Biol. 2016, 23, 838–846. [Google Scholar] [CrossRef]
- He, X.; Du, K.; Wang, Y.; Fan, J.; Li, M.; Ni, D.; Lu, S.; Bian, X.; Liu, Y. Autopromotion of K-Ras4B feedback activation through an SOS-mediated long-range allosteric effect. Front. Mol. Biosci. 2022, 9, 860962. [Google Scholar] [CrossRef] [PubMed]
- Spencer-Smith, R.; O’Bryan, J.P. Direct inhibition of RAS: Quest for the Holy Grail? Semin. Cancer Biol. 2019, 54, 138–148. [Google Scholar] [CrossRef]
- Wang, G.; Bai, Y.; Cui, J.; Zong, Z.; Gao, Y.; Zheng, Z. Computer-aided drug design boosts RAS inhibitor discovery. Molecules 2022, 27, 5710. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Fu, S.; Hao, Q.; Ying, H.; Wang, J.; Shen, T. Multiple medicinal chemistry strategies of targeting KRAS: State-of-the art and future directions. Bioorg. Chem. 2024, 144, 107092. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, D.; Cheng, X. Recent advances in covalent drug discovery. Pharmaceuticals 2023, 16, 663. [Google Scholar] [CrossRef]
- Mahran, R.; Kapp, J.N.; Valtonen, S.; Champagne, A.; Ning, J.; Gillette, W.; Stephen, A.G.; Hao, F.; Plückthun, A.; Härmä, H. Beyond KRAS (G12C): Biochemical and computational characterization of sotorasib and adagrasib binding specificity and the critical role of H95 and Y96. ACS Chem. Biol. 2024, 19, 2152–2164. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS–MAPK reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J. Acquired resistance to KRASG12C inhibition in cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, D.; Zhou, Z.; Wang, H.; Li, Y.; Zheng, H.; Tan, J.; Wu, J.; Yang, Q.; Tian, H. The research advances in Kirsten rat sarcoma viral oncogene homolog (KRAS)-related cancer during 2013 to 2022: A scientometric analysis. Front. Oncol. 2024, 14, 1345737. [Google Scholar] [CrossRef] [PubMed]
- Moghadamchargari, Z.; Shirzadeh, M.; Liu, C.; Schrecke, S.; Packianathan, C.; Russell, D.H.; Zhao, M.; Laganowsky, A. Molecular assemblies of the catalytic domain of SOS with KRas and oncogenic mutants. Proc. Natl. Acad. Sci. USA 2021, 118, e2022403118. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, X.; Wu, C.; Wang, Y.; Zhang, J. Current advances and development strategies of targeting son of sevenless 1 (SOS1) in drug discovery. Eur. J. Med. Chem. 2024, 268, 116282. [Google Scholar] [CrossRef] [PubMed]
- Ikram, S.; Sayyah, E.; Durdağı, S. Identifying Potential SOS1 Inhibitors via Virtual Screening of Multiple Small Molecule Libraries against KRAS-SOS1 Interaction. ChemBioChem 2024, 25, e202400008. [Google Scholar] [CrossRef]
- Srisongkram, T.; Khamtang, P.; Weerapreeyakul, N. Prediction of KRASG12C inhibitors using conjoint fingerprint and machine learning-based QSAR models. J. Mol. Graph. Model. 2023, 122, 108466. [Google Scholar]
- Evelyn, C.R.; Duan, X.; Biesiada, J.; Seibel, W.L.; Meller, J.; Zheng, Y. Rational design of small molecule inhibitors targeting the Ras GEF, SOS1. Chem. Biol. 2014, 21, 1618–1628. [Google Scholar] [PubMed]
- Gomez-Gutierrez, P.; Rubio-Martinez, J.; Perez, J.J. Discovery of Hit Compounds Targeting the P4 Allosteric Site of K-RAS, Identified through Ensemble-Based Virtual Screening. J. Chem. Inf. Model. 2023, 63, 6412–6422. [Google Scholar] [PubMed]
- Chen, T.; Tang, X.; Wang, Z.; Feng, F.; Xu, C.; Zhao, Q.; Wu, Y.; Sun, H.; Chen, Y. Inhibition of son of sevenless homologue 1 (SOS1): Promising therapeutic treatment for KRAS-mutant cancers. Eur. J. Med. Chem. 2023, 261, 115828. [Google Scholar]
- Margarit, S.M.; Sondermann, H.; Hall, B.E.; Nagar, B.; Hoelz, A.; Pirruccello, M.; Bar-Sagi, D.; Kuriyan, J. Structural evidence for feedback activation by Ras· GTP of the Ras-specific nucleotide exchange factor SOS. Cell 2003, 112, 685–695. [Google Scholar] [PubMed]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.-S.; Bar-Sagi, D.; Kuriyan, J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell 2004, 119, 393–405. [Google Scholar]
- Liao, T.-J.; Jang, H.; Fushman, D.; Nussinov, R. Allosteric KRas4B can modulate SOS1 fast and slow Ras activation cycles. Biophys. J. 2018, 115, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H. BI-3406, a potent and selective SOS1–KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Ketcham, J.M.; Haling, J.; Khare, S.; Bowcut, V.; Briere, D.M.; Burns, A.C.; Gunn, R.J.; Ivetac, A.; Kuehler, J.; Kulyk, S. Design and discovery of MRTX0902, a potent, selective, brain-penetrant, and orally bioavailable inhibitor of the SOS1: KRAS protein–protein interaction. J. Med. Chem. 2022, 65, 9678–9690. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.; Prakash, P.; Gorfe, A.A. Computational allosteric ligand binding site identification on Ras proteins. Acta Biochim. Biophys. Sin. 2016, 48, 3–10. [Google Scholar] [CrossRef]
- Marshall, C.B.; KleinJan, F.; Gebregiworgis, T.; Lee, K.Y.; Fang, Z.; Eves, B.J.; Liu, N.F.; Gasmi-Seabrook, G.M.C.; Enomoto, M.; Ikura, M. NMR in integrated biophysical drug discovery for RAS: Past, present, and future. J. Biomol. NMR 2020, 74, 531–554. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef]
- Tatli, O.; Doganay, G.D. Recent developments in targeting ras downstream effectors for ras-driven cancer therapy. Molecules 2021, 26, 7561. [Google Scholar] [CrossRef]
- Negri, F.; Bottarelli, L.; De’ Angelis, G.L.; Gnetti, L. KRAS: A Druggable Target in Colon Cancer Patients. Int. J. Mol. Sci. 2022, 23, 4120. [Google Scholar] [CrossRef]
- Shi, Y.; Zheng, H.; Wang, T.; Zhou, S.; Zhao, S.; Li, M.; Cao, B. Targeting KRAS: From metabolic regulation to cancer treatment. Mol. Cancer 2025, 24, 9. [Google Scholar] [PubMed]
- Kaynak, B.T.; Bahar, I.; Doruker, P. Essential site scanning analysis: A new approach for detecting sites that modulate the dispersion of protein global motions. Comput. Struct. Biotechnol. J. 2020, 18, 1577–1586. [Google Scholar] [PubMed]
- Kurkcuoglu Levitas, A.O. Exploring allosteric communication in multiple states of the bacterial ribosome using residue network analysis. Turkish J. Biol. 2018, 42, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Yuce, M.; Sarica, Z.; Ates, B.; Kurkcuoglu, O. Exploring species-specific inhibitors with multiple target sites on S. aureus pyruvate kinase using a computational workflow. J. Biomol. Struct. Dyn. 2023, 41, 3496–3510. [Google Scholar] [CrossRef]
- Inan, T.; Yuce, M.; MacKerell, A.D., Jr.; Kurkcuoglu, O. Exploring Druggable Binding Sites on the Class A GPCRs Using the Residue Interaction Network and Site Identification by Ligand Competitive Saturation. ACS omega 2024, 9, 40154–40171. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Lukman, S.; Hocker, H.J.; Sayyah, J.; Brown, J.H.; McCammon, J.A.; Gorfe, A.A. Novel allosteric sites on Ras for lead generation. PLoS ONE 2011, 6, e25711. [Google Scholar] [CrossRef]
- Buhrman, G.; Casey, O.; Zerbe, B.; Kearney, B.M.; Napoleon, R.; Kovrigina, E.A.; Vajda, S.; Kozakov, D.; Kovrigin, E.L.; Mattos, C. Analysis of binding site hot spots on the surface of Ras GTPase. J. Mol. Biol. 2011, 413, 773–789. [Google Scholar] [CrossRef]
- McCarthy, M.J.; Pagba, C.V.; Prakash, P.; Naji, A.K.; Van Der Hoeven, D.; Liang, H.; Gupta, A.K.; Zhou, Y.; Cho, K.J.; Hancock, J.F.; et al. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. [Google Scholar] [CrossRef] [PubMed]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef]
- Hall, B.E.; Yang, S.S.; Boriack-Sjodin, P.A.; Kuriyan, J.; Bar-Sagi, D. Structure-based Mutagenesis Reveals Distinct Functions for Ras Switch 1 and Switch 2 in Sos-catalyzed Guanine Nucleotide Exchange. J. Biol. Chem. 2001, 276, 27629–27637. [Google Scholar] [CrossRef]
- Bhadhadhara, K.; Jani, V.; Koulgi, S.; Sonavane, U.; Joshi, R. Studying early structural changes in SOS1 mediated KRAS activation mechanism. Curr. Res. Struct. Biol. 2024, 7, 100115. [Google Scholar] [CrossRef]
- Nastasă, C.; Tamaian, R.; Oniga, O.; Tiperciuc, B. 5-Arylidene (chromenyl-methylene)-thiazolidinediones: Potential new agents against mutant oncoproteins K-Ras, N-Ras and B-Raf in colorectal cancer and melanoma. Medicina 2019, 55, 85. [Google Scholar] [CrossRef]
- Albratty, M.; Alhazmi, H.A. Novel pyridine and pyrimidine derivatives as promising anticancer agents: A review. Arab. J. Chem. 2022, 15, 103846. [Google Scholar] [CrossRef]
- Liew, S.K.; Malagobadan, S.; Arshad, N.M.; Nagoor, N.H. A review of the structure—Activity relationship of natural and synthetic antimetastatic compounds. Biomolecules 2020, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Bergner, A.; Cockcroft, X.; Fischer, G.; Gollner, A.; Hela, W.; Kousek, R.; Mantoulidis, A.; Martin, L.J.; Mayer, M.; Müllauer, B. KRAS binders hidden in nature. Chem. Eur. J. 2019, 25, 12037–12041. [Google Scholar] [CrossRef]
- Al-Muntaser, S.M.; Al-Karmalawy, A.A.; El-Naggar, A.M.; Ali, A.K.; Abd El-Sattar, N.E.A.; Abbass, E.M. Novel 4-thiophenyl-pyrazole, pyridine, and pyrimidine derivatives as potential antitumor candidates targeting both EGFR and VEGFR-2; design, synthesis, biological evaluations, and in silico studies. RSC Adv. 2023, 13, 12184–12203. [Google Scholar] [CrossRef] [PubMed]
- Freitas, L.B.d.O.; Borgati, T.F.; de Freitas, R.P.; Ruiz, A.L.T.G.; Marchetti, G.M.; de Carvalho, J.E.; da Cunha, E.F.F.; Ramalho, T.C.; Alves, R.B. Synthesis and antiproliferative activity of 8-hydroxyquinoline derivatives containing a 1,2,3-triazole moiety. Eur. J. Med. Chem. 2014, 84, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Manish, M.; Mishra, S.; Anand, A.; Subbarao, N. Computational molecular interaction between SARS-CoV-2 main protease and theaflavin digallate using free energy perturbation and molecular dynamics. Comput. Biol. Med. 2022, 150, 106125. [Google Scholar] [CrossRef]
- Abbott, J.R.; Hodges, T.R.; Daniels, R.N.; Patel, P.A.; Kennedy, J.P.; Howes, J.E.; Akan, D.T.; Burns, M.C.; Sai, J.; Sobolik, T. Discovery of aminopiperidine indoles that activate the guanine nucleotide exchange factor SOS1 and modulate RAS signaling. J. Med. Chem. 2018, 61, 6002–6017. [Google Scholar] [PubMed]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [PubMed]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar]
- Jiang, Z.-B.; Wang, W.-J.; Xu, C.; Xie, Y.-J.; Wang, X.-R.; Zhang, Y.-Z.; Huang, J.-M.; Huang, M.; Xie, C.; Liu, P. Luteolin and its derivative apigenin suppress the inducible PD-L1 expression to improve anti-tumor immunity in KRAS-mutant lung cancer. Cancer Lett. 2021, 515, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Kouassi, K.A.R.; Ganiyou, A.; Didier, D.G.G.; Benié, A.; Nahossé, Z. In silico Docking of rhodanine derivatives and 3D-QSAR study to identify potent prostate cancer inhibitors. Comput. Chem. 2022, 10, 19–52. [Google Scholar] [CrossRef]
- Luo, Z.; Paunović, N.; Leroux, J.-C. Physical methods for enhancing drug absorption from the gastrointestinal tract. Adv. Drug Deliv. Rev. 2021, 175, 113814. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.; Vikal, A.; Patel, P.; Narang, R.K.; Kurmi, B. Das Enhancing Oral drug absorption: Overcoming physiological and pharmaceutical barriers for improved bioavailability. AAPS PharmSciTech 2024, 25, 228. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased scale and scope after 10 years of protein dynamics modelling with Python. Bioinformatics 2021, 37, 3657–3659. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [PubMed]
- Yuce, M.; Cicek, E.; Inan, T.; Dag, A.B.; Kurkcuoglu, O.; Sungur, F.A. Repurposing of FDA-approved drugs against active site and potential allosteric drug-binding sites of COVID-19 main protease. Proteins Struct. Funct. Bioinforma. 2021, 89, 1425–1441. [Google Scholar] [CrossRef]
- Yuce, M.; Ates, B.; Yasar, N.I.; Sungur, F.A.; Kurkcuoglu, O. A computational workflow to determine drug candidates alternative to aminoglycosides targeting the decoding center of E. coli ribosome. J. Mol. Graph. Model. 2024, 131, 108817. [Google Scholar] [CrossRef]
- BIOVIA; Dassault Systèmes. Discovery Studio Visualizer, version 2020; Dassault Systèmes: San Diego, CA, USA, 2020.
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84-es. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The nose–hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Alnajjar, R.; Mohamed, N.; Kawafi, N. Bicyclo[1.1.1]Pentane as phenyl substituent in atorvastatin drug to improve physicochemical properties: Drug-likeness, DFT, pharmacokinetics, docking, and molecular dynamic simulation. J. Mol. Struct. 2021, 1230, 129628. [Google Scholar]
- Azam, F.; Eid, E.E.M.; Almutairi, A. Targeting SARS-CoV-2 main protease by teicoplanin: A mechanistic insight by docking, MM/GBSA and molecular dynamics simulation. J. Mol. Struct. 2021, 1246, 131124. [Google Scholar] [PubMed]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar]
- Nussinov, R.; Zhang, M.; Maloney, R.; Jang, H. Ras isoform-specific expression, chromatin accessibility, and signaling. Biophys. Rev. 2021, 13, 489–505. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarica, Z.; Kurkcuoglu, O.; Sungur, F.A. In Silico Identification of Putative Allosteric Pockets and Inhibitors for the KRASG13D-SOS1 Complex in Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 3293. https://doi.org/10.3390/ijms26073293
Sarica Z, Kurkcuoglu O, Sungur FA. In Silico Identification of Putative Allosteric Pockets and Inhibitors for the KRASG13D-SOS1 Complex in Cancer Therapy. International Journal of Molecular Sciences. 2025; 26(7):3293. https://doi.org/10.3390/ijms26073293
Chicago/Turabian StyleSarica, Zehra, Ozge Kurkcuoglu, and Fethiye Aylin Sungur. 2025. "In Silico Identification of Putative Allosteric Pockets and Inhibitors for the KRASG13D-SOS1 Complex in Cancer Therapy" International Journal of Molecular Sciences 26, no. 7: 3293. https://doi.org/10.3390/ijms26073293
APA StyleSarica, Z., Kurkcuoglu, O., & Sungur, F. A. (2025). In Silico Identification of Putative Allosteric Pockets and Inhibitors for the KRASG13D-SOS1 Complex in Cancer Therapy. International Journal of Molecular Sciences, 26(7), 3293. https://doi.org/10.3390/ijms26073293