
101 Machine Learning Algorithms for Mining Esophageal Squamous Cell Carcinoma Neoantigen Prognostic Models in Single-Cell Data
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of Cell Types
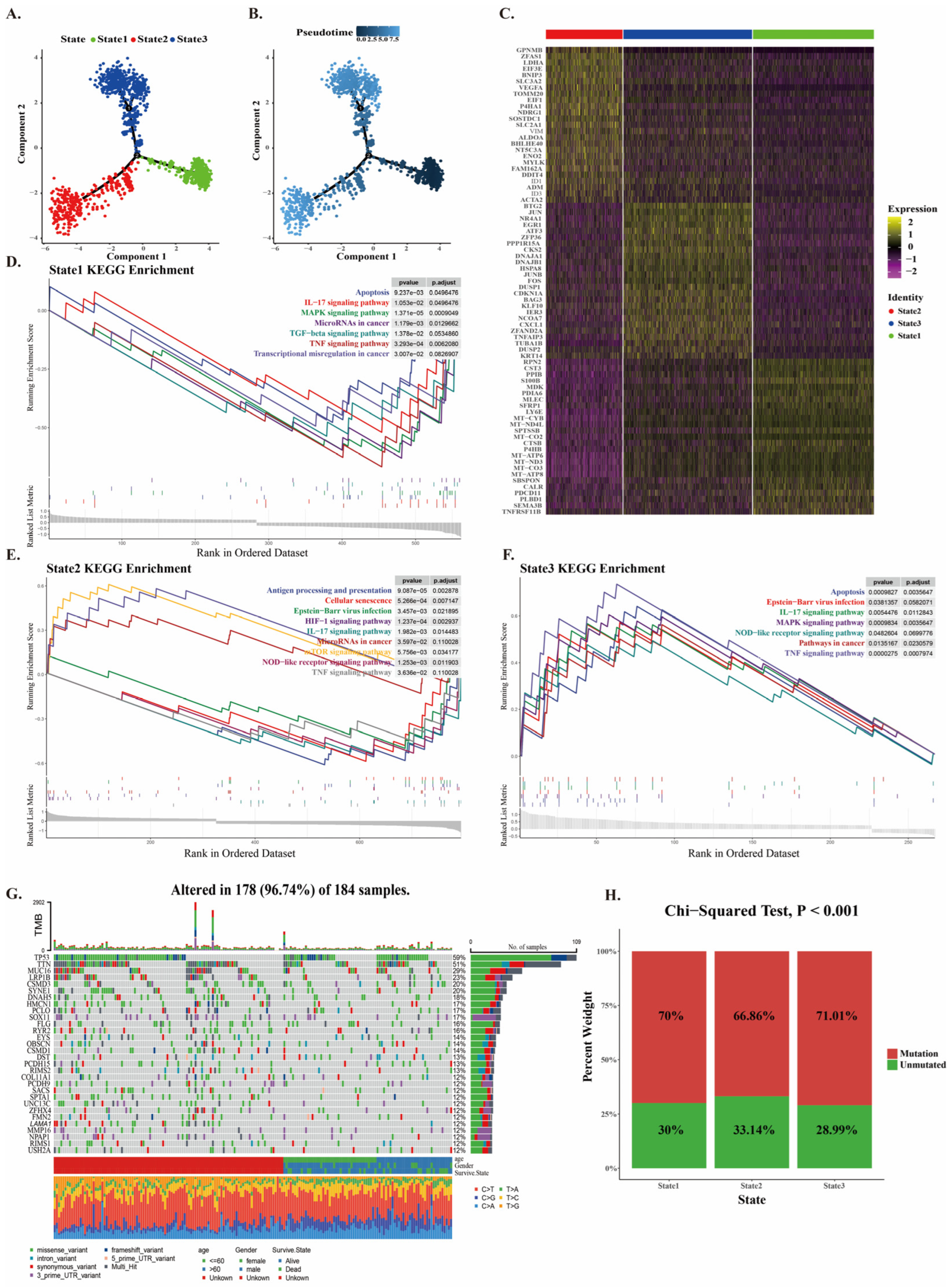
2.2. Mutation of CNV Cancer Cells at Different Developmental Stages
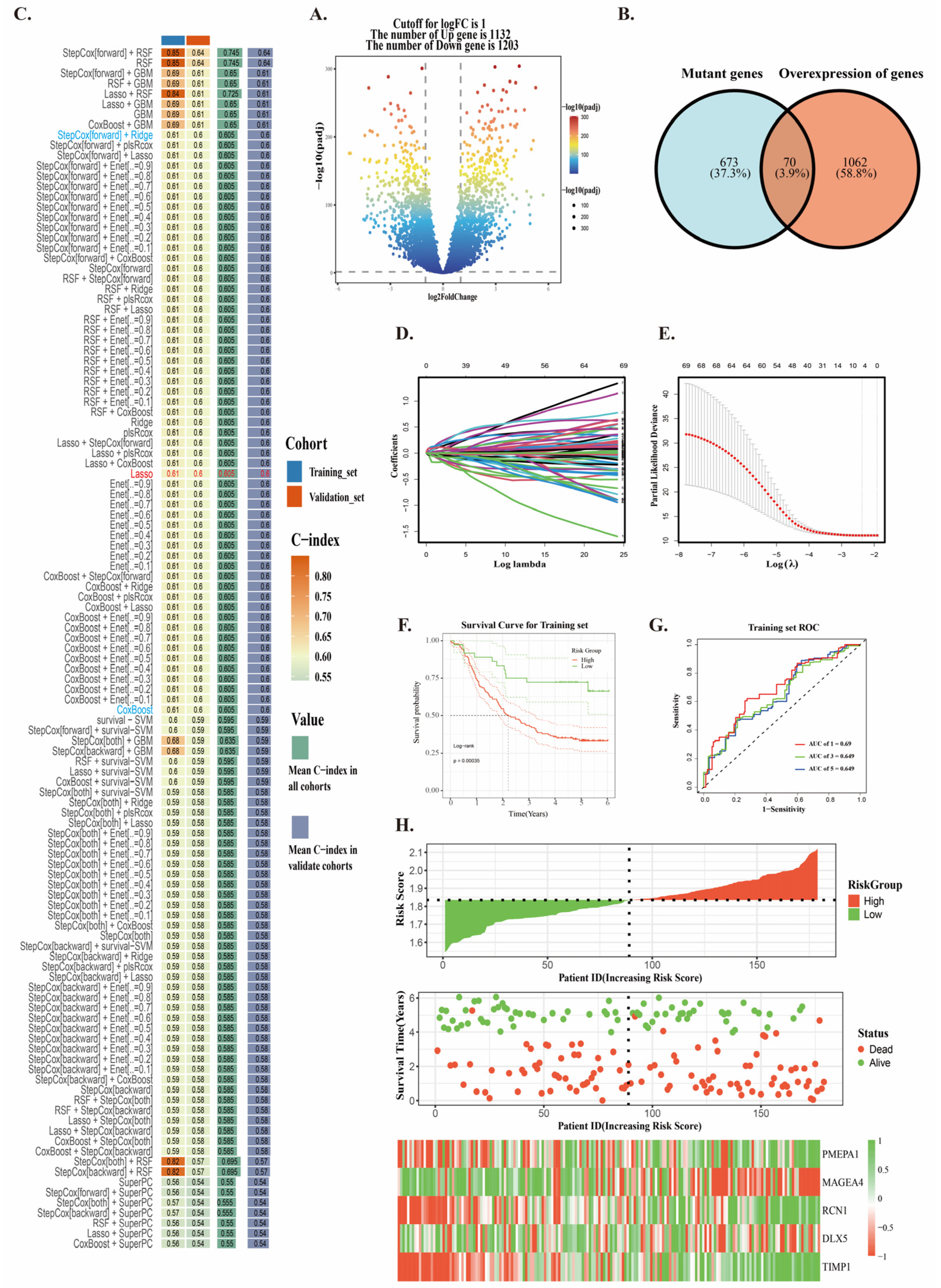
2.3. Construction of Neoantigen Prognostic Risk Model
2.4. The Antigen Presentation Effect and Immune Microenvironment of the Prognostic Risk Model
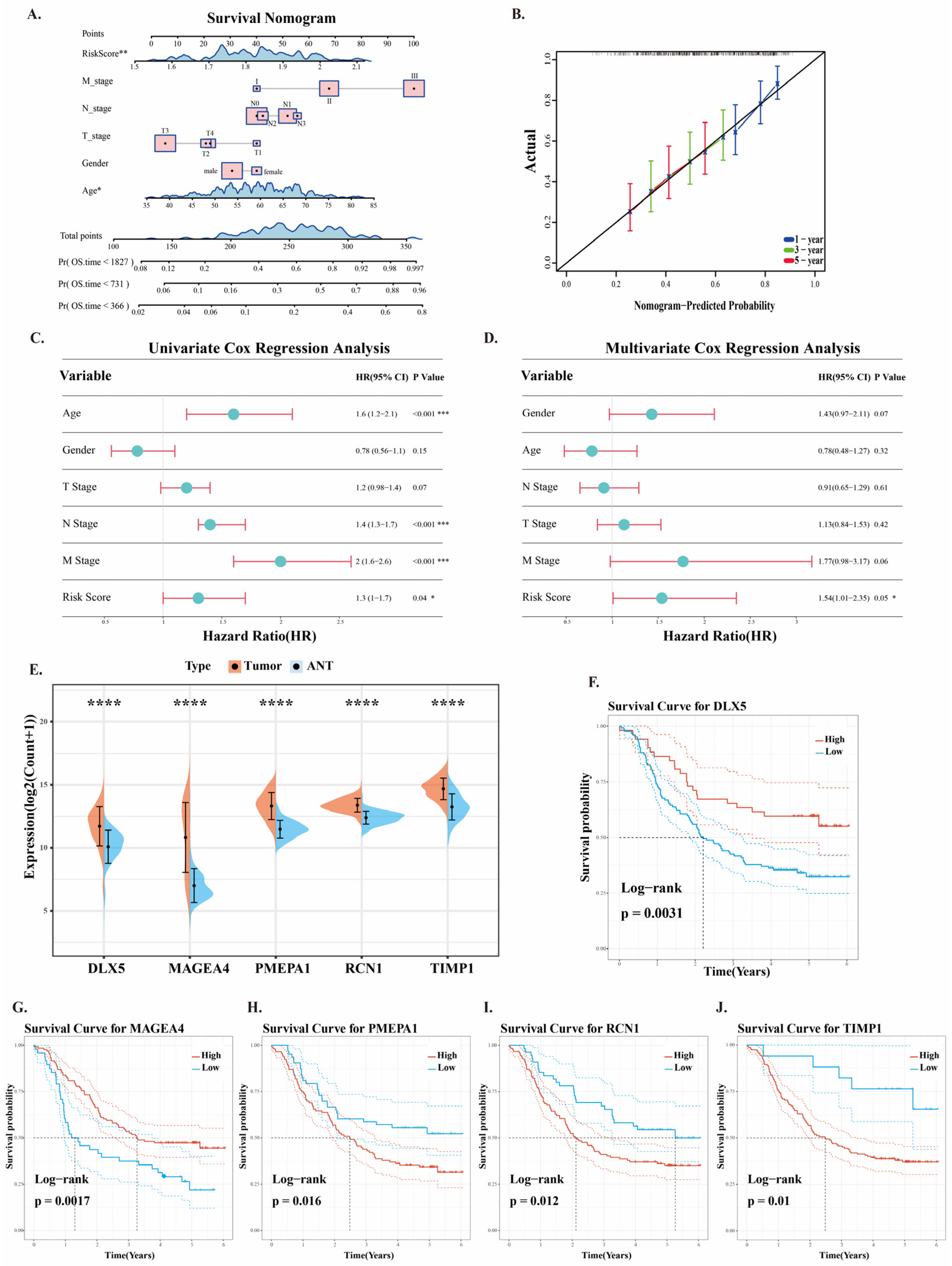
2.5. Clinical Application of Prognostic Risk Models
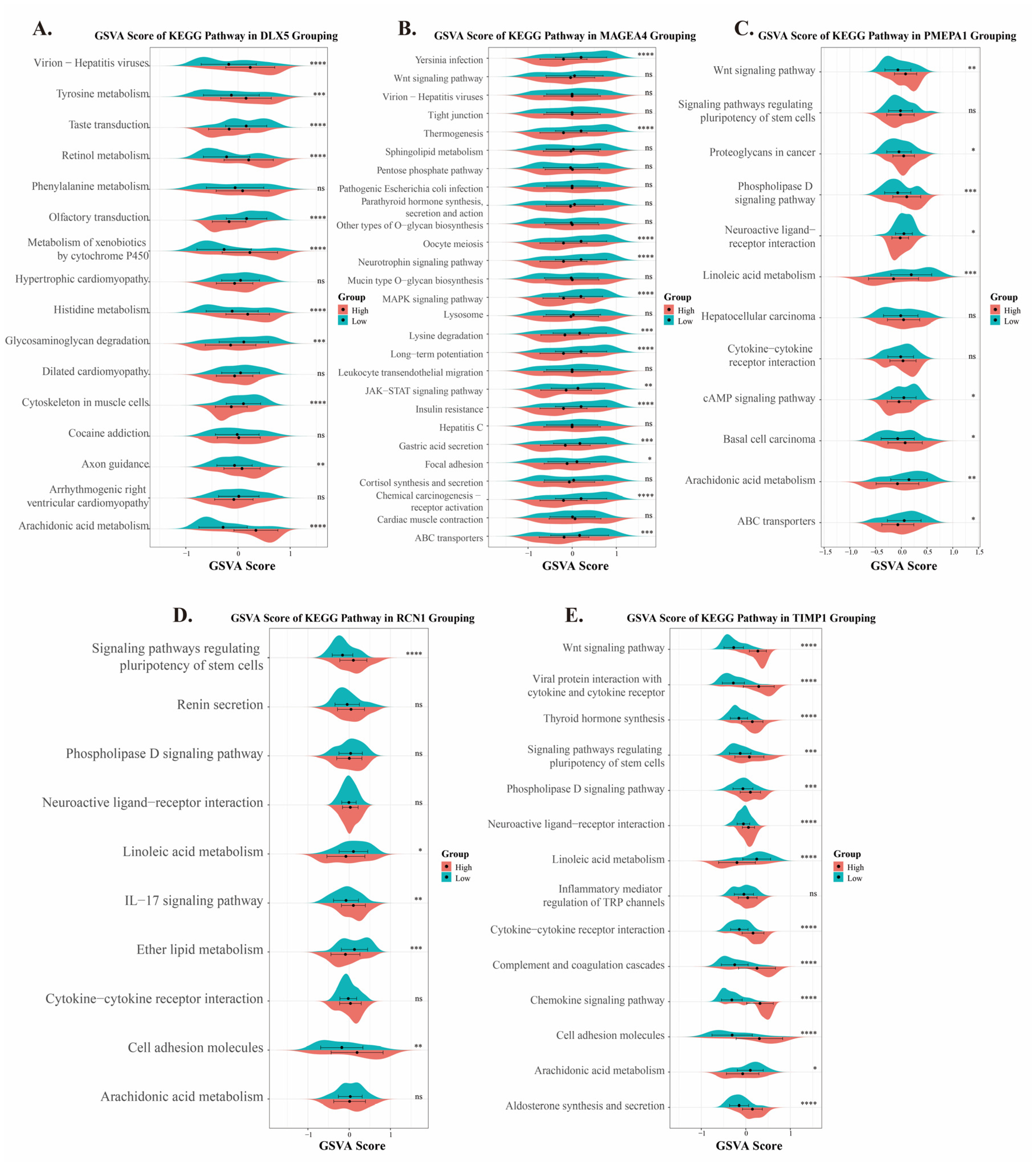
2.6. Analysis of Pathway Functions of Five Prognostic Genes in ESCC
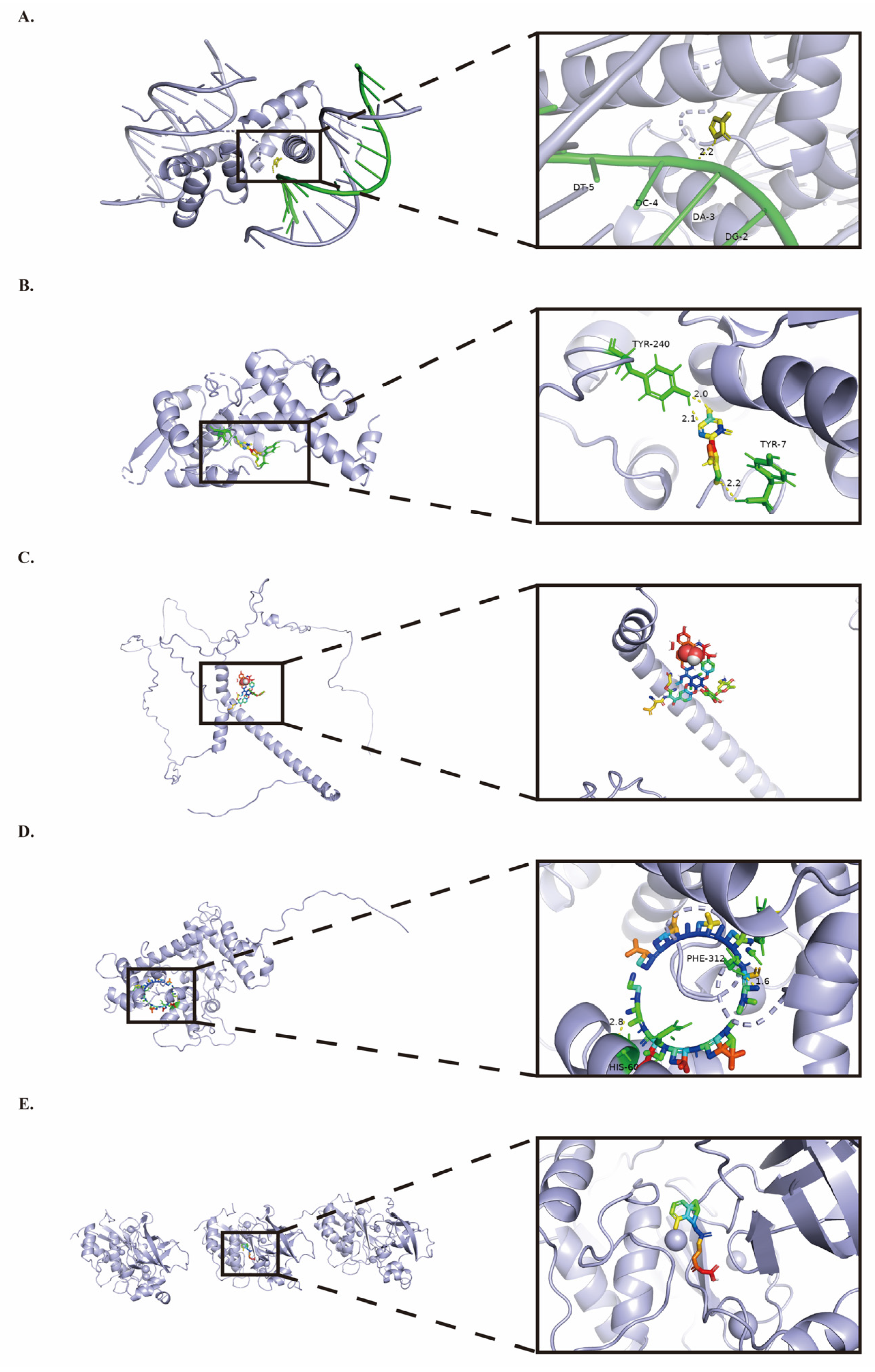
2.7. Prognostic Gene Small Molecule Drug Docking
3. Discussion
4. Materials and Methods
4.1. Data Sources Used for Analysis
4.2. Single-Cell Sequencing Analysis
4.3. The Aneuploid Cell Population in ESCC
4.4. Pseudotime and Somatic Mutation Analysis
4.5. Constructing a Neoantigen Prognostic Risk Model
4.6. Antigen Presentation Effect
4.7. Immune Microenvironment
4.8. Pathway Analysis of Prognostic Genes in High- and Low-Risk Groups
4.9. Small Molecule Drug Screening and Docking
4.10. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ESCC | Esophageal Squamous Cell Carcinoma |
| EAC | Esophageal Adenocarcinoma |
| APC | Antigen-presenting cells |
| DC | Dendritic cells |
| CTL | Cytotoxic T lymphocyte |
| SNV | Single Nucleotide Variant |
| CNV | Copy Number Variation |
| GSEA | Gene Set Enrichment Analysis |
| SuperPC | Supervised Principal Components |
| GBM | Gradient Boosting Machine |
| Survival-SVM | Survival Support Vector Machine |
| Enet | Elastic Net |
| PlsRcox | Partial Least Squares Regression Cox |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| RSF | Random Survival Forest |
| ROC | Receiver Operating Characteristic |
| TME | Tumor Microenvironment |
| GSVA | Gene Set Variation Analysis |
| ssGSEA | Single-sample Gene Set Enrichment Analysis |
| ICI | Immune Checkpoint Inhibition |
| ICP | Immune Checkpoint Proteins |
| OS | Overall Survival |
| MET | Mesenchymal-Epithelial Transition |
| LD | Linear dichroism |
| ESCA | Esophageal carcinoma |
References
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [PubMed]
- Yang, H.; Wang, F.; Hallemeier, C.L.; Lerut, T.; Fu, J. Oesophageal cancer. Lancet 2024, 404, 1991–2005. [Google Scholar] [PubMed]
- Morgan, E.; Soerjomataram, I.; Rumgay, H.; Coleman, H.G.; Thrift, A.P.; Vignat, J.; Laversanne, M.; Ferlay, J.; Arnold, M. The Global Landscape of Esophageal Squamous Cell Carcinoma and Esophageal Adenocarcinoma Incidence and Mortality in 2020 and Projections to 2040: New Estimates From GLOBOCAN 2020. Gastroenterology 2022, 163, 649–658. [Google Scholar]
- Uhlenhopp, D.J.; Uhlenhopp, D.J.; Then, E.O.; Sunkara, T.; Gaduputi, V. Epidemiology of esophageal cancer: Update in global trends, etiology and risk factors. Clin. J. Gastroenterol. 2020, 13, 1010–1021. [Google Scholar]
- Pakzad, R.; Mohammadian-Hafshejani, A.; Khosravi, B.; Soltani, S.; Pakzad, I.; Mohammadian, M.; Salehiniya, H.; Momenimovahed, Z. The incidence and mortality of esophageal cancer and their relationship to development in Asia. Ann. Transl. Med. 2016, 4, 29. [Google Scholar]
- Puhr, H.C.; Prager, G.W.; Ilhan-Mutlu, A. How we treat esophageal squamous cell carcinoma. ESMO Open 2023, 8, 100789. [Google Scholar]
- Shoji, Y.; Koyanagi, K.; Kanamori, K.; Tajima, K.; Ogimi, M.; Ninomiya, Y.; Yamamoto, M.; Kazuno, A.; Nabeshima, K.; Nishi, T.; et al. Immunotherapy for esophageal cancer: Where are we now and where can we go. World J. Gastroenterol. 2024, 30, 2496–2501. [Google Scholar]
- Lybaert, L.; Lefever, S.; Fant, B.; Smits, E.; De Geest, B.; Breckpot, K.; Dirix, L.; Feldman, S.A.; van Criekinge, W.; Thielemans, K.; et al. Challenges in neoantigen-directed therapeutics. Cancer Cell 2023, 41, 15–40. [Google Scholar]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar]
- Esprit, A.; de Mey, W.; Bahadur Shahi, R.; Thielemans, K.; Franceschini, L.; Breckpot, K. Neo-Antigen mRNA Vaccines. Vaccines 2020, 8, 776. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, H.; Tian, L.; Pang, Z.; Yang, Q.; Huang, T.; Fan, J.; Song, L.; Tong, Y.; Fan, H. COVID-19 vaccine development: Milestones, lessons and prospects. Signal Transduct. Target. Ther. 2022, 7, 146. [Google Scholar] [PubMed]
- Sayour, E.J.; Boczkowski, D.; Mitchell, D.A.; Nair, S.K. Cancer mRNA vaccines: Clinical advances and future opportunities. Nat. Rev. Clin. Oncol. 2024, 21, 489–500. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Vishweshwaraiah, Y.L.; Dokholyan, N.V. mRNA vaccines for cancer immunotherapy. Front. Immunol. 2022, 13, 1029069. [Google Scholar]
- Huang, X.; Zhang, G.; Tang, T.Y.; Gao, X.; Liang, T.B. Personalized pancreatic cancer therapy: From the perspective of mRNA vaccine. Mil. Med. Res. 2022, 9, 53. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, L.; Luo, Y.; Zhang, S.; Pu, Y.; Chen, Y.; Guo, W.; Yao, J.; Shao, M.; Fan, W.; et al. Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat. Commun. 2021, 12, 5291. [Google Scholar]
- Huang, F.L.; Yu, S.J. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J. Surg. 2018, 41, 210–215. [Google Scholar]
- Cha, J.H.; Chan, L.C.; Song, M.S.; Hung, M.C. New Approaches on Cancer Immunotherapy. Cold Spring Harb. Perspect. Med. 2020, 10, a036863. [Google Scholar]
- Haen, S.; Löffler, M.W.; Rammensee, H.G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [PubMed]
- Huang, X.; Zhang, G.; Tang, T.; Liang, T. Identification of tumor antigens and immune subtypes of pancreatic adenocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 44. [Google Scholar] [PubMed]
- Huang, X.; Tang, T.; Zhang, G.; Liang, T. Identification of tumor antigens and immune subtypes of cholangiocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 50. [Google Scholar] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar]
- Yuan, X.; Wu, H.; Han, N.; Xu, H.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Notch signaling and EMT in non-small cell lung cancer: Biological significance and therapeutic application. J. Hematol. Oncol. 2014, 7, 87. [Google Scholar]
- Meisel, C.T.; Porcheri, C.; Mitsiadis, T.A. Cancer Stem Cells, Quo Vadis? The Notch Signaling Pathway in Tumor Initiation and Progression. Cells 2020, 9, 1879. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar]
- Ren, H.Z.; Pan, G.Q.; Wang, J.S.; Wen, J.F.; Wang, K.S.; Luo, G.Q.; Shan, X.Z. Reduced stratifin expression can serve as an independent prognostic factor for poor survival in patients with esophageal squamous cell carcinoma. Dig. Dis. Sci. 2010, 55, 2552–2560. [Google Scholar]
- Chang, J.; Zhao, X.; Wang, Y.; Liu, T.; Zhong, C.; Lao, Y.; Zhang, S.; Liao, H.; Bai, F.; Lin, D.; et al. Genomic alterations driving precancerous to cancerous lesions in esophageal cancer development. Cancer Cell 2023, 41, 2038–2050. [Google Scholar]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar]
- Sharad, S.; Dobi, A.; Srivastava, S.; Srinivasan, A.; Li, H. PMEPA1 Gene Isoforms: A Potential Biomarker and Therapeutic Target in Prostate Cancer. Biomolecules 2020, 10, 1221. [Google Scholar] [CrossRef] [PubMed]
- Saadi, A.; Shannon, N.B.; Lao-Sirieix, P.; O’Donovan, M.; Walker, E.; Clemons, N.J.; Hardwick, J.S.; Zhang, C.; Das, M.; Save, V.; et al. Stromal genes discriminate preinvasive from invasive disease, predict outcome, and highlight inflammatory pathways in digestive cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 2177–2182. [Google Scholar] [PubMed]
- Zhang, Y.; Zhu, H.; Sun, N.; Zhang, X.; Liang, G.; Zhu, J.; Xia, L.; Kou, Y.; Lu, J. Linc00941 regulates esophageal squamous cell carcinoma via functioning as a competing endogenous RNA for miR-877-3p to modulate PMEPA1 expression. Aging 2021, 13, 17830–17846. [Google Scholar] [PubMed]
- Guo, H.; Shu, J.; Hu, G.; Liu, B.; Li, J.; Sun, J.; Wang, X.; Liu, H.; Xiong, S.; Tang, Y.; et al. Downregulation of RCN1 inhibits esophageal squamous cell carcinoma progression and M2 macrophage polarization. PLoS ONE 2024, 19, e0302780. [Google Scholar]
- Liu, H.; Guo, H.; Wu, Y.; Hu, Q.; Hu, G.; He, H.; Yin, Y.; Nan, X.; Lin, G.; Han, J.; et al. RCN1 deficiency inhibits oral squamous cell carcinoma progression and THP-1 macrophage M2 polarization. Sci. Rep. 2023, 13, 21488. [Google Scholar]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar]
- Kozłowski, M.; Laudański, W.; Mroczko, B.; Szmitkowski, M.; Milewski, R.; Łapuć, G. Serum tissue inhibitor of metalloproteinase 1 (TIMP-1) and vascular endothelial growth factor A (VEGF-A) are associated with prognosis in esophageal cancer patients. Adv. Med. Sci. 2013, 58, 227–234. [Google Scholar]
- Griffith-Jones, S.; Álvarez, L.; Mukhopadhyay, U.; Gharbi, S.; Rettel, M.; Adams, M.; Hennig, J.; Bhogaraju, S. Structural basis for RAD18 regulation by MAGEA4 and its implications for RING ubiquitin ligase binding by MAGE family proteins. EMBO J. 2024, 43, 1273–1300. [Google Scholar]
- Chen, X.; Cai, S.; Wang, L.; Zhang, X.; Li, W.; Cao, X. Analysis of the function of MAGE-A in esophageal carcinoma by bioinformatics. Medicine 2019, 98, e15774. [Google Scholar]
- Levi, G.; de Lombares, C.; Giuliani, C.; Iannuzzi, V.; Aouci, R.; Garagnani, P.; Franceschi, C.; Grimaud-Hervé, D.; Narboux-Nême, N. DLX5/6 GABAergic Expression Affects Social Vocalization: Implications for Human Evolution. Mol. Biol. Evol. 2021, 38, 4748–4764. [Google Scholar]
- Brooks, Y.S.; Ostano, P.; Jo, S.H.; Dai, J.; Getsios, S.; Dziunycz, P.; Hofbauer, G.F.; Cerveny, K.; Chiorino, G.; Lefort, K.; et al. Multifactorial ERβ and NOTCH1 control of squamous differentiation and cancer. J. Clin. Investig. 2014, 124, 2260–2276. [Google Scholar] [PubMed]
- Huang, Y.; Yang, Q.; Zheng, Y.; Lin, L.; Xu, X.; Xu, X.E.; Silva, T.C.; Hazawa, M.; Peng, L.; Cao, H.; et al. Activation of bivalent factor DLX5 cooperates with master regulator TP63 to promote squamous cell carcinoma. Nucleic Acids Res. 2021, 49, 9246–9263. [Google Scholar] [PubMed]
- Hong, S.; Zhang, Z.; Liu, H.; Tian, M.; Zhu, X.; Zhang, Z.; Wang, W.; Zhou, X.; Zhang, F.; Ge, Q.; et al. B Cells Are the Dominant Antigen-Presenting Cells that Activate Naive CD4(+) T Cells upon Immunization with a Virus-Derived Nanoparticle Antigen. Immunity 2018, 49, 695–708. [Google Scholar]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar]
- Cooper, D.S. Antithyroid drugs. N. Engl. J. Med. 2005, 352, 905–917. [Google Scholar]
- Farooqi, A.S.; Hong, J.Y.; Cao, J.; Lu, X.; Price, I.R.; Zhao, Q.; Kosciuk, T.; Yang, M.; Bai, J.J.; Lin, H. Novel Lysine-Based Thioureas as Mechanism-Based Inhibitors of Sirtuin 2 (SIRT2) with Anticancer Activity in a Colorectal Cancer Murine Model. J. Med. Chem. 2019, 62, 4131–4141. [Google Scholar]
- Viswas, R.S.; Pundir, S.; Lee, H. Design and synthesis of 4-piperazinyl quinoline derived urea/thioureas for anti-breast cancer activity by a hybrid pharmacophore approach. J. Enzym. Inhib. Med. Chem. 2019, 34, 620–630. [Google Scholar]
- Li, X.; Li, Y.; Dong, L.; Chang, Y.; Zhang, X.; Wang, C.; Chen, M.; Bo, X.; Chen, H.; Han, W.; et al. Decitabine priming increases anti-PD-1 antitumor efficacy by promoting CD8+ progenitor exhausted T cell expansion in tumor models. J. Clin. Investig. 2023, 133, e165673. [Google Scholar]
- Garcia-Manero, G.; McCloskey, J.; Griffiths, E.A.; Yee, K.W.L.; Zeidan, A.M.; Al-Kali, A.; Deeg, H.J.; Patel, P.A.; Sabloff, M.; Keating, M.M.; et al. Oral decitabine-cedazuridine versus intravenous decitabine for myelodysplastic syndromes and chronic myelomonocytic leukaemia (ASCERTAIN): A registrational, randomised, crossover, pharmacokinetics, phase 3 study. Lancet Haematol. 2024, 11, e15–e26. [Google Scholar]
- Jeffres, M.N. The Whole Price of Vancomycin: Toxicities, Troughs, and Time. Drugs 2017, 77, 1143–1154. [Google Scholar] [PubMed]
- Survase, S.A.; Kagliwal, L.D.; Annapure, U.S.; Singhal, R.S. Cyclosporin A—A review on fermentative production, downstream processing and pharmacological applications. Biotechnol. Adv. 2011, 29, 418–435. [Google Scholar]
- di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar]
- Brown, G. Targeting the Retinoic Acid Pathway to Eradicate Cancer Stem Cells. Int. J. Mol. Sci. 2023, 24, 2373. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar]
- Wang, Z.; Jensen, M.A.; Zenklusen, J.C. A Practical Guide to The Cancer Genome Atlas (TCGA). Methods Mol. Biol. 2016, 1418, 111–141. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar]
- Reel, S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar]
- Ma, Y.; Jin, J.; Xue, Z.; Zhao, J.; Cai, W.; Zhang, W. Integrated multi-omics analysis and machine learning developed a prognostic model based on mitochondrial function in a large multicenter cohort for Gastric Cancer. J. Transl. Med. 2024, 22, 381. [Google Scholar]
- Tian, D.; Yan, H.J.; Huang, H.; Zuo, Y.J.; Liu, M.Z.; Zhao, J.; Wu, B.; Shi, L.Z.; Chen, J.Y. Machine Learning-Based Prognostic Model for Patients After Lung Transplantation. JAMA Netw. Open 2023, 6, e2312022. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, X.; Ren, J.; Zhang, Z.; Ju, H.; Diao, X.; Jiang, S.; Zhang, J. Glycosyltransferase-related prognostic and diagnostic biomarkers of uterine corpus endometrial carcinoma. Comput. Biol. Med. 2023, 163, 107164. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Wiegers, T.C.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2023. Nucleic Acids Res. 2023, 51, D1257–D1262. [Google Scholar]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Tang, Y.; Qi, Q.; Pang, J.; Chen, Y.; Wang, H.; Liang, J.; Tang, W. 101 Machine Learning Algorithms for Mining Esophageal Squamous Cell Carcinoma Neoantigen Prognostic Models in Single-Cell Data. Int. J. Mol. Sci. 2025, 26, 3373. https://doi.org/10.3390/ijms26073373
Sun Y, Tang Y, Qi Q, Pang J, Chen Y, Wang H, Liang J, Tang W. 101 Machine Learning Algorithms for Mining Esophageal Squamous Cell Carcinoma Neoantigen Prognostic Models in Single-Cell Data. International Journal of Molecular Sciences. 2025; 26(7):3373. https://doi.org/10.3390/ijms26073373
Chicago/Turabian StyleSun, Yingjie, Yuheng Tang, Qi Qi, Jianyu Pang, Yongzhi Chen, Hui Wang, Jiaxiang Liang, and Wenru Tang. 2025. "101 Machine Learning Algorithms for Mining Esophageal Squamous Cell Carcinoma Neoantigen Prognostic Models in Single-Cell Data" International Journal of Molecular Sciences 26, no. 7: 3373. https://doi.org/10.3390/ijms26073373
APA StyleSun, Y., Tang, Y., Qi, Q., Pang, J., Chen, Y., Wang, H., Liang, J., & Tang, W. (2025). 101 Machine Learning Algorithms for Mining Esophageal Squamous Cell Carcinoma Neoantigen Prognostic Models in Single-Cell Data. International Journal of Molecular Sciences, 26(7), 3373. https://doi.org/10.3390/ijms26073373