
Abdominal Aortic Aneurysm and Liver Fibrosis: Clinical Evidence and Molecular Pathomechanisms




Abstract
:1. Introduction
2. Epidemiology and Risk Factors
2.1. Methodological Limitations
2.2. Liver Fibrosis Due to MASLD
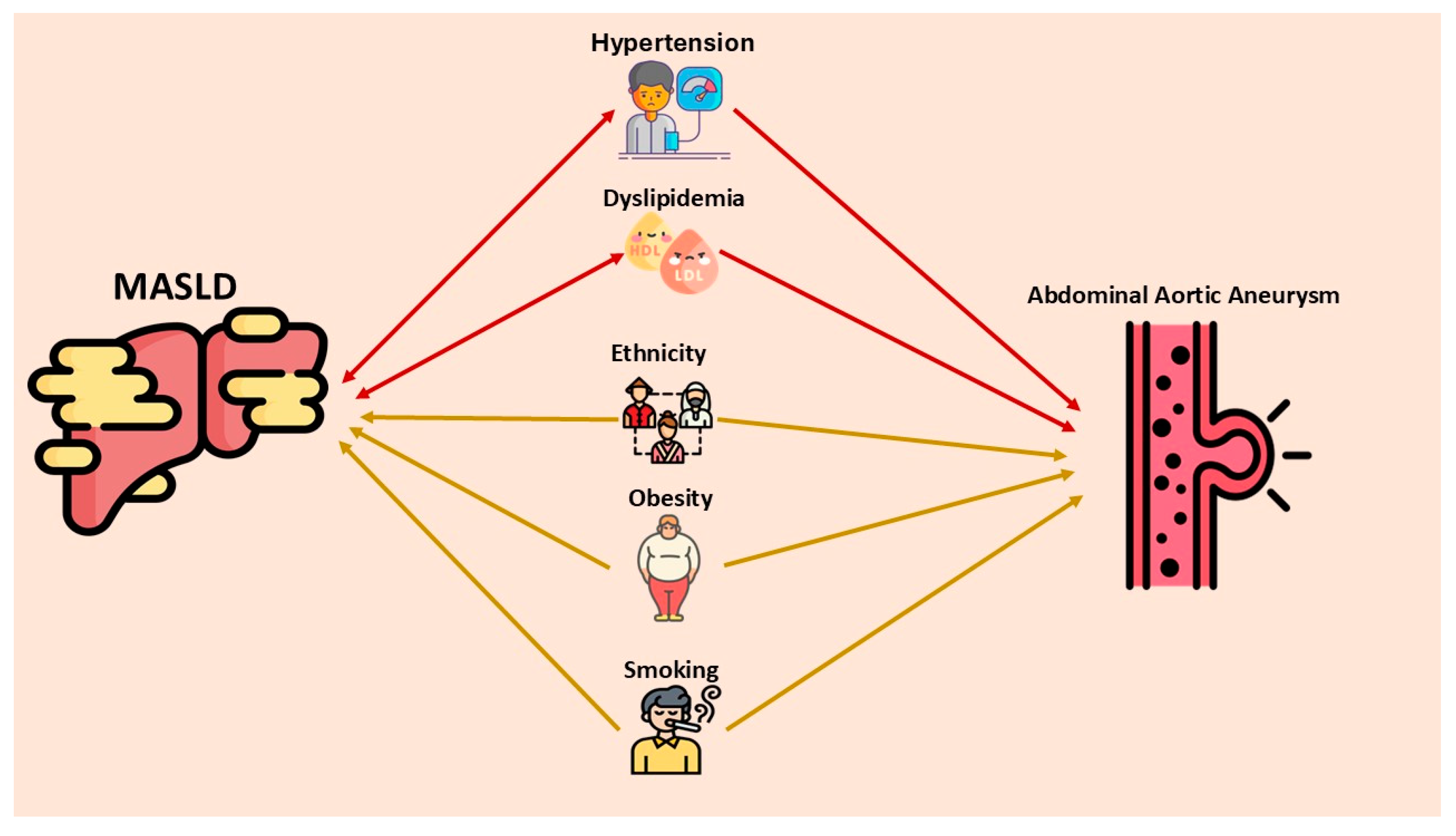
2.3. Shared Risk Factors Between AAA and MASLD
2.3.1. Hypercholesterolemia
2.3.2. Smoking
2.3.3. Obesity
2.3.4. Diabetes
2.3.5. Arterial Hypertension
2.3.6. Aortic Distensibility Is Altered and Endothelial Dysfunction Occurs in MASLD
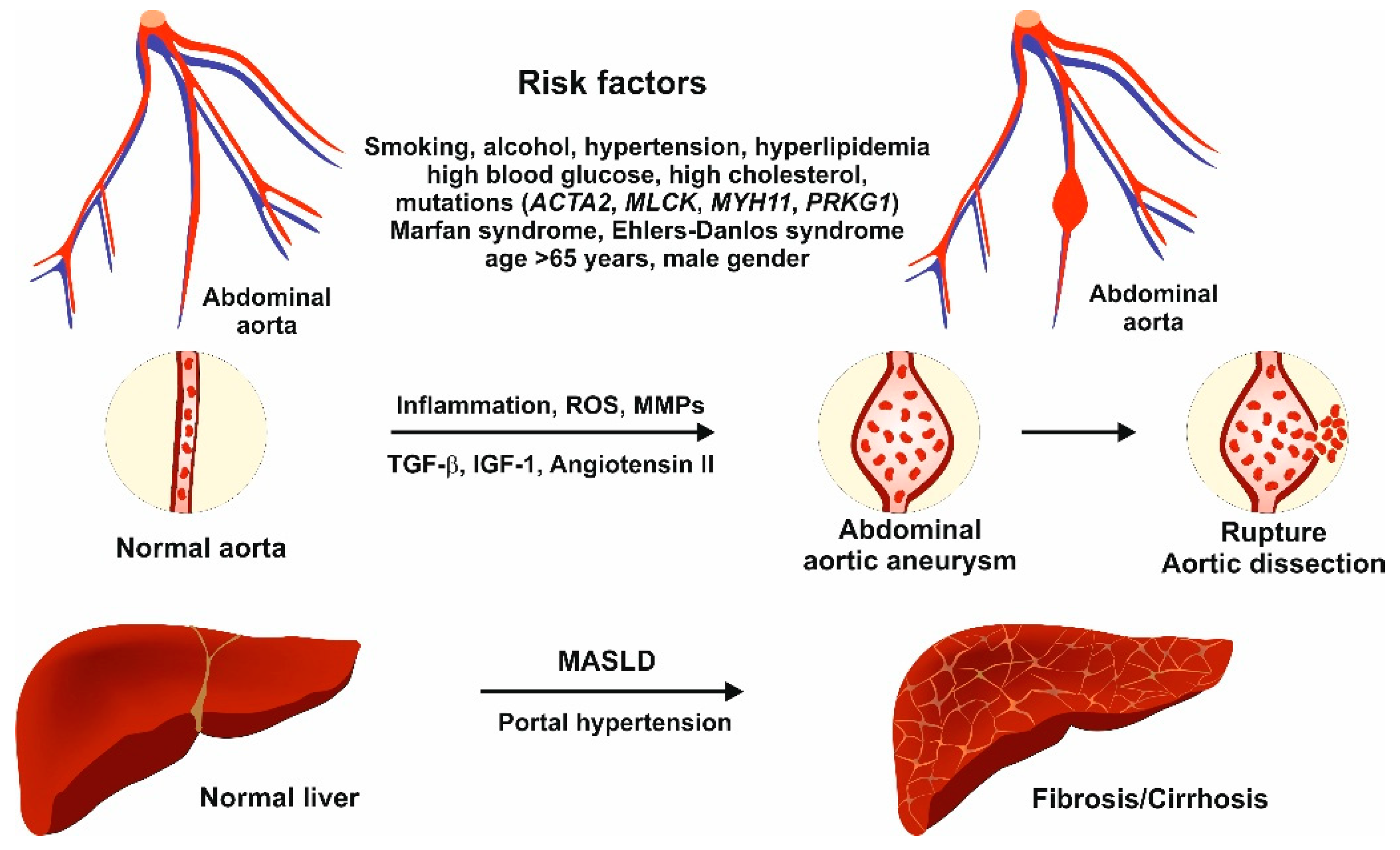
3. Molecular Pathomechanisms
3.1. Pathomechanisms of Abdominal Aortic Aneurysm
3.2. Gene Mutations in the Pathogenesis of Abdominal Aortic Aneurysm
3.3. Relation Between Abdominal Aortic Aneurysm and Liver Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA | abdominal aortic aneurysm |
| CKM | cardiovascular–kidney–metabolic |
| CLD | chronic liver disease |
| ECM | extracellular matrix |
| EVAR | endovascular aneurysm repair |
| HSC(s) | hepatic stellate cell(s) |
| HTN | arterial hypertension |
| MASH | metabolic dysfunction-associated steatohepatitis |
| MASLD | metabolic dysfunction-associated steatotic liver disease |
| MS | Marfan syndrome |
| MMP(s) | matrix metalloproteinase(s) |
| RLC | regulatory light chain |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| SLD | steatotic liver disease |
| SMC(s) | smooth muscle cell(s) |
| T2DM | type 2 diabetes mellitus |
| TIMP(s) | tissue inhibitor of metalloproteinase(s) |
References
- Golledge, J. Abdominal aortic aneurysm: Update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225–242. [Google Scholar] [CrossRef]
- Calgi, M.P.; McNeil, J.S. Abdominal Aortic Aneurysms (Etiology, Epidemiology, and Natural History). Anesthesiol. Clin. 2022, 40, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Kessler, V.; Klopf, J.; Eilenberg, W.; Neumayer, C.; Brostjan, C. AAA Revisited: A Comprehensive Review of Risk Factors, Management, and Hallmarks of Pathogenesis. Biomedicines 2022, 10, 94. [Google Scholar] [CrossRef]
- Zecca, F.; Faa, G.; Sanfilippo, R.; Saba, L. How to improve epidemiological trustworthiness concerning abdominal aortic aneurysms. Vascular 2024, 17085381241257747. [Google Scholar] [CrossRef] [PubMed]
- Körfer, D.; Erhart, P.; Dihlmann, S.; Hakimi, M.; Böckler, D.; Peters, A.S. Histopathological Characterization of Abdominal Aortic Aneurysms from Patients with Multiple Aneurysms Compared to Patients with a Single Abdominal Aortic Aneurysm. Biomedicines 2023, 11, 1311. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gao, J.; Wu, J.; Zheng, Y. The Impact of COVID-19 Infection on Abdominal Aortic Aneurysms: Mechanisms and Clinical Implications. Cardiovasc. Ther. 2024, 2024, 7288798. [Google Scholar] [CrossRef]
- Jamalinia, M.; Lonardo, A.; Weiskirchen, R. Sex and Gender Differences in Liver Fibrosis: Pathomechanisms and Clinical Outcomes. Fibrosis 2024, 2, 10006. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Baffy, G.; Weiskirchen, R. Liver fibrosis as a barometer of systemic health by gauging the risk of extrahepatic disease. Metab. Target Organ Damage 2024, 4, 41. [Google Scholar] [CrossRef]
- Bagheri Lankarani, K.; Jamalinia, M.; Zare, F.; Heydari, S.T.; Ardekani, A.; Lonardo, A. Liver-Kidney-Metabolic Health, Sex, and Menopause Impact Total Scores and Monovessel vs. Multivessel Coronary Artery Calcification. Adv. Ther. 2025. [CrossRef]
- Ndumele, C.E.; Neeland, I.J.; Tuttle, K.R.; Chow, S.L.; Mathew, R.O.; Khan, S.S.; Coresh, J.; Baker-Smith, C.M.; Carnethon, M.R.; Després, J.P.; et al. A Synopsis of the Evidence for the Science and Clinical Management of Cardiovascular-Kidney-Metabolic (CKM) Syndrome: A Scientific Statement From the American Heart Association. Circulation 2023, 148, 1636–1664. [Google Scholar] [CrossRef]
- Stefan, N.; Lonardo, A.; Targher, G. Role of steatotic liver disease in prediction and prevention of cardiometabolic diseases. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Peshkova, I.O.; Schaefer, G.; Koltsova, E.K. Atherosclerosis and aortic aneurysm—Is inflammation a common denominator? FEBS J. 2016, 283, 1636–1652. [Google Scholar] [CrossRef]
- Toghill, B.J.; Saratzis, A.; Bown, M.J. Abdominal aortic aneurysm-an independent disease to atherosclerosis? Cardiovasc. Pathol. 2017, 27, 71–75. [Google Scholar] [CrossRef]
- Wang, X.; Sun, J.; Chang, N.; Liu, M.; Zhang, S. Association between non-alcoholic fatty liver disease and progression of abdominal aortic aneurysm: A multicenter study. BMC Med. Imaging 2025, 25, 24. [Google Scholar] [CrossRef]
- Takayama, T.; Miyata, T.; Nagawa, H. True abdominal aortic aneurysm in Marfan syndrome. J. Vasc. Surg. 2009, 49, 1162–1165. [Google Scholar] [CrossRef]
- Marrocco-Trischitta, M.M.; Kahlberg, A.; Astore, D.; Tshiombo, G.; Mascia, D.; Chiesa, R. Outcome in cirrhotic patients after elective surgical repair of infrarenal aortic aneurysm. J. Vasc. Surg. 2011, 53, 906–911. [Google Scholar] [CrossRef]
- Mahamid, M.; Khoury, T.; Mahamid, B.; Sbeit, W.; Mari, A.; Nseir, W. The interplay between abdominal aortic aneurysm, metabolic syndrome and fatty liver disease: A retrospective case-control study. Diabetes Metab. Syndr. Obes. 2019, 12, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Zettervall, S.L.; Dansey, K.; Swerdlow, N.J.; Soden, P.; Evenson, A.; Schermerhorn, M.L. Aspartate transaminase to platelet ratio index and Model for End-Stage Liver Disease scores are associated with morbidity and mortality after endovascular aneurysm repair among patients with liver dysfunction. J. Vasc. Surg. 2020, 72, 904–909. [Google Scholar] [CrossRef]
- Jia, Y.; Li, Y.; Yu, J.; Jiang, W.; Liu, Y.; Zeng, R.; Wan, Z.; Liao, X.; Li, D.; Zhao, Q. Association between metabolic dysfunction-associated fatty liver disease and abdominal aortic aneurysm. Nutr. Metab. Cardiovasc. Dis. 2024, 34, 953–962. [Google Scholar] [CrossRef]
- Jamalinia, M.; Mirhosseini, S.A.; Ranjbar, M.; Bagheri Lankarani, K.; Hosseinzadeh, A. Association of liver fibrosis with aneurysm size and mortality risk in patients undergoing open abdominal aortic aneurysm repair. Sci. Rep. 2025, 15, 3301. [Google Scholar] [CrossRef]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210.e2. [Google Scholar] [CrossRef] [PubMed]
- Barshes, N.R.; Bidare, D.; Kougias, P.; Mills, J.L., Sr.; LeMaire, S.A. Racial and ethnic disparities in abdominal aortic aneurysm evaluation and treatment rates in Texas. J. Vasc. Surg. 2022, 76, 141–148.e1. [Google Scholar] [CrossRef] [PubMed]
- Loufopoulos, G.; Tasoudis, P.; Koudounas, G.; Zoupas, I.; Madouros, N.; Sá, M.P.; Karkos, C.D.; Giannopoulos, S.; Tassiopoulos, A.K. Long-Term Outcomes of Open Versus Endovascular Treatment for Abdominal Aortic Aneurysm: Systematic Review and Meta-Analysis With Reconstructed Time-to-Event Data. J. Endovasc. Ther. 2023, 15266028231204805. [Google Scholar] [CrossRef]
- Ou, T.Y.; Huy, L.D.; Mayne, J.; Shih, C.L.; Mai Xuan, H.; Thi Hong Nguyen, N.; Nguyen Hoai, L.; Thi My Bui, L.; Chang, Y.M.; Abdi, A.A.; et al. Global mortality of chronic liver diseases attributable to Hepatitis B virus and Hepatitis C virus infections from 1990 to 2019 and projections to 2030. J. Infect. Public Health 2024, 17, 102443. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, X.D.; Shapiro, M.D.; Lip, G.Y.H.; Tilg, H.; Valenti, L.; Somers, V.K.; Byrne, C.D.; Targher, G.; Yang, W.; et al. Global burden of metabolic diseases, 1990–2021. Metabolism 2024, 160, 155999. [Google Scholar] [CrossRef]
- Braunwald, E. From cardiorenal to cardiovascular-kidney-metabolic syndromes. Eur. Heart J. 2025, 46, 682–684. [Google Scholar] [CrossRef]
- Pham, M.H.C.; Sigvardsen, P.E.; Fuchs, A.; Kühl, J.T.; Sillesen, H.; Afzal, S.; Nordestgaard, B.G.; Køber, L.V.; Kofoed, K.F. Aortic aneurysms in a general population cohort: Prevalence and risk factors in men and women. Eur. Heart J. Cardiovasc. Imaging 2024, 25, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Agache, P.; Fanian, F. Body Surface Area. In Agache’s Measuring the Skin; Humbert, P., Maibach, H., Fanian, F., Agache, P., Eds.; Springer: Cham, Switzerland, 2015. [Google Scholar] [CrossRef]
- Liu, J.; Sawada, H.; Howatt, D.A.; Moorleghen, J.J.; Vsevolozhskaya, O.; Daugherty, A.; Lu, H.S. Hypercholesterolemia Accelerates Both the Initiation and Progression of Angiotensin II-induced Abdominal Aortic Aneurysms. Ann. Vasc. Med. Res. 2020, 6, 1099. [Google Scholar]
- Ioannou, G.N.; Landis, C.S.; Jin, G.Y.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated With Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv. Exp. Med. Biol. 2018, 1061, 19–44. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.F.; Armenian, H.K.; Friesen, P.P. Risk factors for abdominal aortic aneurysm: Results of a case-control study. Am. J. Epidemiol. 2000, 151, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Forsdahl, S.H.; Singh, K.; Solberg, S.; Jacobsen, B.K. Risk factors for abdominal aortic aneurysms: A 7-year prospective study: The Tromsø Study, 1994–2001. Circulation 2009, 119, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.H.L.; Nguyen, V.M.; Adermark, L.; Alvarez, G.G.; Shelley, D.; Ng, N. Factors Influencing Tobacco Smoking and Cessation Among People Living with HIV: A Systematic Review and Meta-analysis. AIDS Behav. 2024, 28, 1858–1881. [Google Scholar] [CrossRef]
- Ekpenyong, M.S.; Jagun, H.; Stephen, H.A.; Bakre, A.T.; Odejimi, O.; Miller, E.; Nyashanu, M.; Bosun-Arije, S.F. Investigation of the prevalence and factors influencing tobacco and alcohol use among adolescents in Nigeria: A systematic literature review. Drug Alcohol. Depend. 2024, 256, 111091. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Schlesinger, S.; Norat, T.; Riboli, E. Tobacco smoking and the risk of abdominal aortic aneurysm: A systematic review and meta-analysis of prospective studies. Sci. Rep. 2018, 8, 14786. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Y.; Feng, Z.; Chen, H. Cigarette Smoke Contributes to the Progression of MASLD: From the Molecular Mechanisms to Therapy. Cells 2025, 14, 221. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; He, Y.; Adeloye, D.; Zhu, Y.; Ye, X.; Yi, Q.; Rahimi, K.; Rudan, I.; Global Health Epidemiology Research Group (GHERG). The Global and Regional Prevalence of Abdominal Aortic Aneurysms: A Systematic Review and Modeling Analysis. Ann. Surg. 2023, 277, 912–919. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef]
- Tao, M.; Yu, P.; Nguyen, B.T.; Mizrahi, B.; Savion, N.; Kolodgie, F.D.; Virmani, R.; Hao, S.; Ozaki, C.K.; Schneiderman, J. Locally applied leptin induces regional aortic wall degeneration preceding aneurysm formation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 311–320. [Google Scholar] [CrossRef]
- Martínez-Martínez, E.; Souza-Neto, F.V.; Jiménez-González, S.; Cachofeiro, V. Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest. Antioxidants 2021, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.; Bianco, R.; Di Gregoli, K.; Johnson, J.L. The contribution of matrix metalloproteinases and their inhibitors to the development, progression, and rupture of abdominal aortic aneurysms. Front. Cardiovasc. Med. 2023, 10, 1248561. [Google Scholar] [CrossRef]
- Larsen, K.L.; Kavaliunaite, E.; Rasmussen, L.M.; Hallas, J.; Diederichsen, A.; Steffensen, F.H.; Busk, M.; Frost, L.; Urbonaviciene, G.; Lambrechtsen, J.; et al. The association between diabetes and abdominal aortic aneurysms in men: Results of two Danish screening studies, a systematic review, and a meta-analysis of population-based screening studies. BMC Cardiovasc. Disord. 2023, 23, 139. [Google Scholar] [CrossRef]
- Dewangga, R.; Winston, K.; Ilhami, L.G.; Indriani, S.; Siddiq, T.; Adiarto, S. Association of metformin use with abdominal aortic aneurysm: A systematic review and meta-analysis. Asian Cardiovasc. Thorac. Ann. 2024, 32, 148–156. [Google Scholar] [CrossRef]
- Kristensen, K.L.; Dahl, M.; Rasmussen, L.M.; Lindholt, J.S. Glycated Hemoglobin Is Associated With the Growth Rate of Abdominal Aortic Aneurysms: A Substudy From the VIVA (Viborg Vascular) Randomized Screening Trial. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 730–736. [Google Scholar] [CrossRef]
- Picatoste, B.; Cerro-Pardo, I.; Blanco-Colio, L.M.; Martín-Ventura, J.L. Protection of diabetes in aortic abdominal aneurysm: Are antidiabetics the real effectors? Front. Cardiovasc. Med. 2023, 10, 1112430. [Google Scholar] [CrossRef]
- Dattani, N.; Sayers, R.D.; Bown, M.J. Diabetes mellitus and abdominal aortic aneurysms: A review of the mechanisms underlying the negative relationship. Diab Vasc. Dis. Res. 2018, 15, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kobeissi, E.; Hibino, M.; Pan, H.; Aune, D. Blood pressure, hypertension and the risk of abdominal aortic aneurysms: A systematic review and meta-analysis of cohort studies. Eur. J. Epidemiol. 2019, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D. Mechanisms of Vascular Remodeling in Hypertension. Am. J. Hypertens. 2021, 34, 432–441. [Google Scholar] [CrossRef]
- Banerjee, P.; Gaddam, N.; Chandler, V.; Chakraborty, S. Oxidative Stress-Induced Liver Damage and Remodeling of the Liver Vasculature. Am. J. Pathol. 2023, 193, 1400–1414. [Google Scholar] [CrossRef]
- Liu, J.; Lv, H.; Wang, J.; Zhu, Q.; Chen, G.; Jiang, Y.; Zhao, K.; Shao, L.; Shi, J.; Pan, X. Blood pressure stratification for predicting liver fibrosis risk in metabolic dysfunction associated fatty liver disease. Ann. Hepatol. 2023, 28, 100892. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.S.; Wentworth, B.J. The Renin-Angiotensin System in Liver Disease. Int. J. Mol. Sci. 2024, 25, 5807. [Google Scholar] [CrossRef]
- Nesci, A.; Ruggieri, V.; Manilla, V.; Spinelli, I.; Santoro, L.; Di Giorgio, A.; Santoliquido, A.; Ponziani, F.R. Endothelial Dysfunction and Liver Cirrhosis: Unraveling of a Complex Relationship. Int. J. Mol. Sci. 2024, 25, 12859. [Google Scholar] [CrossRef] [PubMed]
- Işilak, Z.; Aparci, M.; Kardeşoğlu, E.; Yiğiner, O.; Uz, O.; Sildiroglu, O.; Ozmen, N.; Yalçin, M.; Cingözbay, B.Y.; Cebeci, B.S. Abnormal aortic elasticity in patients with liver steatosis. Diabetes Res. Clin. Pract. 2010, 87, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Manesis, E.; Baou, K.; Papatheodoridis, G.; Koskinas, J.; Tiniakos, D.; Aznaouridis, K.; Archimandritis, A.; Stefanadis, C. Increased arterial stiffness and impaired endothelial function in nonalcoholic Fatty liver disease: A pilot study. Am. J. Hypertens. 2010, 23, 1183–1189. [Google Scholar] [CrossRef]
- Li, X.; Shi, H.; Wang, Z.; Chang, L.; Zhang, M.; Dong, X. Arterial stiffness is increased in nondiabetic, nonhypertensive postmenopausal women with nonalcoholic fatty liver disease. J. Hypertens. 2017, 35, 1226–1234. [Google Scholar] [CrossRef]
- Gentili, A.; Daviddi, G.; De Vuono, S.; Ricci, M.A.; Di Filippo, F.; Alaeddin, A.; Mannarino, M.R.; Boni, M.; Vaudo, G.; Lupattelli, G. Non-alcoholic fatty liver disease fibrosis score and preclinical vascular damage in morbidly obese patients. Dig. Liver Dis. 2016, 48, 904–908. [Google Scholar] [CrossRef]
- Al-Hamoudi, W.; Alsadoon, A.; Hassanian, M.; Alkhalidi, H.; Abdo, A.; Nour, M.; Halwani, R.; Sanai, F.; Alsharaabi, A.; Alswat, K.; et al. Endothelial dysfunction in nonalcoholic steatohepatitis with low cardiac disease risk. Sci. Rep. 2020, 10, 8825. [Google Scholar] [CrossRef]
- Arinc, H.; Sarli, B.; Baktir, A.O.; Saglam, H.; Demirci, E.; Dogan, Y.; Kurtul, S.; Karaman, H.; Erden, A.; Karaman, A. Serum gamma glutamyl transferase and alanine transaminase concentrations predict endothelial dysfunction in patients with non-alcoholic steatohepatitis. Ups. J. Med. Sci. 2013, 118, 228–234. [Google Scholar] [CrossRef]
- Lonardo, A.; Ndrepepa, J. Concise review: Gamma-glutamyl transferase—Evolution from an indiscriminate liver test to a biomarker of cardiometabolic risk. Metab. Target Organ Damage 2022, 2, 17. [Google Scholar] [CrossRef]
- Calanchini, M.; Moolla, A.; Tomlinson, J.W.; Cobbold, J.F.; Grossman, A.; Fabbri, A.; Turner, H.E. Liver biochemical abnormalities in Turner syndrome: A comprehensive characterization of an adult population. Clin. Endocrinol. 2018, 89, 667–676. [Google Scholar] [CrossRef]
- Twohig, P.; Li, L.; Danford, D.; Craft, M.; Yetman, A.T. Prevalence of hepatic steatosis and fibrosis in Turner syndrome: A prospective case-control study. Liver Int. 2024, 44, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Maham Yuan, Z.; Lu, Y.; Wei, J.; Wu, J.; Yang, J.; Cai, Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front. Immunol. 2021, 11, 609161. [Google Scholar] [CrossRef]
- Márquez-Sánchez, A.C.; Koltsova, E.K. Immune and inflammatory mechanisms of abdominal aortic aneurysm. Front. Immunol. 2022, 13, 989933. [Google Scholar] [CrossRef]
- Kazaleh, M.; Gioscia-Ryan, R.; Ailawadi, G.; Salmon, M. Oxidative Stress and the Pathogenesis of Aortic Aneurysms. Biomedicines 2023, 12, 3. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, R.; Oo, A.Y.; Xiao, Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharmaceuticals 2019, 12, 118. [Google Scholar] [CrossRef]
- Baas, A.F.; Medic, J.; van ’t Slot, R.; de Kovel, C.G.; Zhernakova, A.; Geelkerken, R.H.; Kranendonk, S.E.; van Sterkenburg, S.M.; Grobbee, D.E.; Boll, A.P.; et al. Association of the TGF-beta receptor genes with abdominal aortic aneurysm. Eur. J. Hum. Genet. 2010, 18, 240–244. [Google Scholar] [CrossRef]
- Biros, E.; Norman, P.E.; Jones, G.T.; van Rij, A.M.; Yu, G.; Moxon, J.V.; Blankensteijn, J.D.; van Sterkenburg, S.M.; Morris, D.; Baas, A.F.; et al. Meta-analysis of the association between single nucleotide polymorphisms in TGF-β receptor genes and abdominal aortic aneurysm. Atherosclerosis 2011, 219, 218–223. [Google Scholar] [CrossRef]
- IJpma, A.; Te Riet, L.; van de Luijtgaarden, K.M.; van Heijningen, P.M.; Burger, J.; Majoor-Krakauer, D.; Rouwet, E.V.; Essers, J.; Verhagen, H.J.M.; van der Pluijm, I. Inflammation and TGF-β Signaling Differ between Abdominal Aneurysms and Occlusive Disease. J. Cardiovasc. Dev. Dis. 2019, 6, 38. [Google Scholar] [CrossRef]
- Stepien, K.L.; Bajdak-Rusinek, K.; Fus-Kujawa, A.; Kuczmik, W.; Gawron, K. Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2022, 23, 11078. [Google Scholar] [CrossRef]
- Lu, H.; Du, W.; Ren, L.; Hamblin, M.H.; Becker, R.C.; Chen, Y.E.; Fan, Y. Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. J. Am. Heart Assoc. 2021, 10, e023601. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Matt, P.; Schoenhoff, F.; Habashi, J.; Holm, T.; Van Erp, C.; Loch, D.; Carlson, O.D.; Griswold, B.F.; Fu, Q.; De Backer, J.; et al. Circulating transforming growth factor-beta in Marfan syndrome. Circulation 2009, 120, 526–532. [Google Scholar] [CrossRef]
- Matt, P.; Habashi, J.; Carrel, T.; Cameron, D.E.; Van Eyk, J.E.; Dietz, H.C. Recent advances in understanding Marfan syndrome: Should we now treat surgical patients with losartan? J. Thorac. Cardiovasc. Surg. 2008, 135, 389–394. [Google Scholar] [CrossRef]
- Lareyre, F.; Clément, M.; Raffort, J.; Pohlod, S.; Patel, M.; Esposito, B.; Master, L.; Finigan, A.; Vandestienne, M.; Stergiopulos, N.; et al. TGFβ (Transforming Growth Factor-β) Blockade Induces a Human-Like Disease in a Nondissecting Mouse Model of Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2171–2181. [Google Scholar] [CrossRef]
- Renard, M.; Callewaert, B.; Baetens, M.; Campens, L.; MacDermot, K.; Fryns, J.P.; Bonduelle, M.; Dietz, H.C.; Gaspar, I.M.; Cavaco, D.; et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int. J. Cardiol. 2013, 165, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Boelman, M.B.; Hansen, T.V.O.; Smith, M.N.; Hammer-Hansen, S.; Christensen, A.H.; Diness, B.R. Aortic dissection in a young male with persistent ductus arteriosus and a novel variant in MYLK. Am. J. Med. Genet. A 2024, 194, e63458. [Google Scholar] [CrossRef]
- Regalado, E.S.; Guo, D.C.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef]
- Guo, D.C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493, Erratum in Nat. Genet. 2008, 40, 255. [Google Scholar] [CrossRef]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Østergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Adès, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A. 2010, 152A, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhou, X.; Jiang, X.; Sun, T. Deletion of ACTA2 in mice promotes angiotensin II induced pathogenesis of thoracic aortic aneurysms and dissections. J. Thorac. Dis. 2018, 10, 4733–4740. [Google Scholar] [CrossRef]
- Wallace, S.E.; Regalado, E.S.; Gong, L.; Janda, A.L.; Guo, D.C.; Russo, C.F.; Kulmacz, R.J.; Hanna, N.; Jondeau, G.; Boileau, C.; et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 2019, 21, 144–151. [Google Scholar] [CrossRef]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]
- Wang, L.; Guo, D.C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.Q.; Zhu, M.S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707, Erratum in Am. J. Hum. Genet. 2011, 88, 516. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, P.; Li, Z.; Shi, Y.; Huang, J.; Li, S.; Liu, Y.; Zhang, Z.; Wang, Y.; Zhu, W.; et al. The Effect of Myosin Light Chain Kinase on the Occurrence and Development of Intracranial Aneurysm. Front. Cell Neurosci. 2018, 12, 416. [Google Scholar] [CrossRef]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462, Erratum in Hum. Mol. Genet. 2008, 17, 158. [Google Scholar] [CrossRef]
- Katz, A.E.; Yang, M.L.; Levin, M.G.; Tcheandjieu, C.; Mathis, M.; Hunker, K.; Blackburn, S.; Eliason, J.L.; Coleman, D.M.; Fendrikova-Mahlay, N.; et al. Fibromuscular Dysplasia and Abdominal Aortic Aneurysms Are Dimorphic Sex-Specific Diseases With Shared Complex Genetic Architecture. Circ. Genom. Precis. Med. 2022, 15, e003496. [Google Scholar] [CrossRef]
- Zhu, B.; Zhao, G.; Witte, D.P.; Hui, D.Y.; Fagin, J.A. Targeted overexpression of IGF-I in smooth muscle cells of transgenic mice enhances neointimal formation through increased proliferation and cell migration after intraarterial injury. Endocrinology 2001, 142, 3598–3606. [Google Scholar] [CrossRef]
- Orstavik, S.; Natarajan, V.; Taskén, K.; Jahnsen, T.; Sandberg, M. Characterization of the human gene encoding the type I alpha and type I beta cGMP-dependent protein kinase (PRKG1). Genomics 1997, 42, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Han, Q.; Liu, Z.; Zhou, W.; Cao, Q.; Zhou, W. Exome sequencing reveals a de novo PRKG1 mutation in a sporadic patient with aortic dissection. BMC Med. Genet. 2018, 19, 218. [Google Scholar] [CrossRef]
- Klarin, D.; Verma, S.S.; Judy, R.; Dikilitas, O.; Wolford, B.N.; Paranjpe, I.; Levin, M.G.; Pan, C.; Tcheandjieu, C.; Spin, J.M.; et al. Genetic Architecture of Abdominal Aortic Aneurysm in the Million Veteran Program. Circulation 2020, 142, 1633–1646. [Google Scholar] [CrossRef]
- van de Luijtgaarden, K.M.; Heijsman, D.; Maugeri, A.; Weiss, M.M.; Verhagen, H.J.; IJpma, A.; Brüggenwirth, H.T.; Majoor-Krakauer, D. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum. Genet. 2015, 134, 881–893. [Google Scholar] [CrossRef]
- Schon, H.T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141, Erratum in Free Radic. Biol. Med. 2021, 162, 174. https://doi.org/10.1016/j.freeradbiomed.2020.06.011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Lin, W.; Yang, B.; Liu, Y.; Huang, W.; Xie, N.; Yang, F.; Lin, Z.; Hu, Z.; Luo, S.; et al. Remnant cholesterol and risk of aortic aneurysm and dissection: A prospective cohort Study from the UK biobank study and mendelian randomization analysis. Lipids Health Dis. 2025, 24, 53. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef]
- Wang, Q.; Yesitayi, G.; Liu, B.; Siti, D.; Ainiwan, M.; Aizitiaili, A.; Ma, X. Targeting metabolism in aortic aneurysm and dissection: From basic research to clinical applications. Int. J. Biol. Sci. 2023, 19, 3869–3891. [Google Scholar] [CrossRef] [PubMed]
- Koba, A.; Yamagishi, K.; Sairenchi, T.; Noda, H.; Irie, F.; Takizawa, N.; Tomizawa, T.; Iso, H.; Ota, H. Risk Factors for Mortality From Aortic Aneurysm and Dissection: Results from a 26-Year Follow-Up of a Community-Based Population. J. Am. Heart Assoc. 2023, 12, e027045. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Vega, M.; Massó, F.; Páez, A.; Vargas-Alarcón, G.; Coral-Vázquez, R.; Mas-Oliva, J.; Carreón-Torres, E.; Pérez-Méndez, Ó. HDL-Mediated Lipid Influx to Endothelial Cells Contributes to Regulating Intercellular Adhesion Molecule (ICAM)-1 Expression and eNOS Phosphorylation. Int. J. Mol. Sci. 2018, 19, 3394. [Google Scholar] [CrossRef] [PubMed]
- Argraves, K.M.; Argraves, W.S. HDL serves as a S1P signaling platform mediating a multitude of cardiovascular effects. J. Lipid Res. 2007, 48, 2325–2333. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Author Year [Ref.] | Origin | Methods | Findings | Conclusions |
|---|---|---|---|---|
| Marrocco-Trischitta J, 2011 [16] | Italy | Between January 2001 and March 2006, 1189 consecutive patients underwent elective open repair of infrarenal AAA, with 24 (2%) of them having cirrhosis. The patients with cirrhosis included 23 males and 1 female with a mean age of 68 ± 7 years. They were retrospectively stratified based on the CTP and MELD scores. Operative variables, perioperative complications, and survival rates were compared to those of 48 matched non-cirrhotic controls, and the impact of CTP and MELD scores on midterm survival was assessed in cirrhotic patients using the Kaplan–Meier log-rank method. | Major perioperative complications occurred to a similar extent among cirrhotic patients and controls. However, the duration of surgery, requirements of intraoperative blood transfusion and duration of hospital stay were all higher in patients with cirrhosis (p = 0.007; p = 0.040; p < 0.0001, respectively). CTP class B cirrhotic subjects had higher requirements for intraoperative blood transfusions (p = 0.029). 2-year actuarial survival rates were 77.4% among patients with cirrhosis vs. 97.8% in controls (log-rank test, p = 0.026). CTP class B and a MELD score ≥10 were associated with reduced mid-term survival rates (p < 0.0001 and p = 0.021, respectively). | Although elective AAA open repair in patients with relatively compensated cirrhosis was safely performed, the reduced life expectancy of patients with cirrhosis and a MELD score ≥10 suggest avoiding this procedure in this specific patient cohort. |
| Mahamid M, 2019 [17] | Israel | A retrospective case-control study was conducted with 495 AAA patients and 500 age- and sex-matched controls. | The prevalence of FLD was higher among AAA patients than in controls (48.9% vs. 21.2% p < 0.005). LRA after adjusting for confounding factors showed that AAA (men: OR 1.29, 95% CI 1.17, 1.49, p = 0.001; women: OR 1.23, 95% CI 1.06, 1.43, p = 0.002), obesity (men: OR 1.32, 95% CI 1.17, 1.59, p < 0.001; women: OR 1.32, 95% CI 1.07, 1.52, p = 0.012), hypertension (men: OR 1.23, 95% CI 1.13, 1.46, p = 0.001; women: OR 1.13, 95% CI 1.00, 1.33, p = 0.045), MS (men: OR 1.31, 95% CI 1.19, 1.53, p = 0.001; women: OR 1.28, 95% CI 1.16, 1.42, p = 0.002) were associated with NAFLD/NASH. The prevalence of liver cirrhosis was 1.23%; and subjects with obesity, diabetes, hypertension, and AAA had an increased risk of cirrhosis (OR 1.89, 95% CI 1.18, 3.22, p = 0.014; OR 1.27, 95% CI 1.09, 2.72, p = 0.0027; OR 2.08, 95% CI 1.29, 3.42, p = 0.004; OR 1.73, 95% CI 1.08, 2.87, p = 0.027, respectively). | Patients with AAA are at an increased risk of NAFLD/NASH, which may progress to cirrhosis. |
| Zettervall SL, 2020 [18] | USA | The National Surgical Quality Improvement Program evaluated all nonemergent EVARs from 2005 to 2016. An APRI >0.5 was used to identify significant liver fibrosis, and demographics, comorbidities, and 30-day outcomes were compared between patients with and without fibrosis. Further analysis was performed to evaluate the effect of increasing MELD scores on 30-day outcomes, using MVRA to adjust for baseline differences. | EVAR was performed in 18,484 patients (2286 with liver fibrosis and 16,198 without). Patients with liver fibrosis had an increased 30-day mortality (1.5% vs. 2.4%; p < 0.01) and significantly higher rates of major morbidities. At MVA, patients with liver fibrosis had a significant increase in 30-day mortality (OR, 1.5; 95%, CI, 1.1–2.1), re-operation (OR, 1.5; 95% CI, 1.2–1.8), pulmonary complications (OR, 1.6; 95% CI, 1.2–2.0), transfusion (OR, 1.7; 95% CI, 1.5–2.0), and discharge other than home (OR, 1.5; 95% CI, 1.3–1.8). Mortality also increased in parallel with an increase in MELD score (MELD <10, 1.3%; MELD 10–15, 2.3%; MELD >15, 4.7%; p < 0.01), and so did major complications (MELD <10, 7%; MELD 10–15, 11%; MELD >15, 15%; p < 0.01). These increases persisted in adjusted analysis. | Liver fibrosis is significantly associated with increased mortality and major morbidity after EVAR. The APRI and MELD scores can be used for preoperative risk stratification. Current 30-day morbidity and mortality rates among patients with MELD scores >10 exceed 5%, which is higher than the annual rupture risk for aneurysms <6 cm, supporting the utilization of an increased size threshold of >6 cm before EVAR in patients with liver fibrosis. |
| Jia Y, 2024 [19] | China | A total of 370,203 participants (36.4% with MAFLD) from the prospective UK Biobank cohort study were followed up for 12.5 years. MAFLD was defined as HS plus metabolic abnormality, T2DM, or overweight/obesity. AAA data was collected using ICD-10 code, and Cox regression was used to analyze the association between MAFLD and AAA. | During follow-up, 1561 (0.4%) of participants developed AAA. In fully adjusted analysis, MAFLD was associated with a significantly higher likelihood of AAA (HR 1.521, 95% CI 1.351–1.712, p < 0.001), and the AAA risk increased with the severity of MAFLD fibrosis scores irrespective of sex, weight, alcohol consumption, and PRS. However, these associations were weaker in the elderly or diabetics (p for interaction <0.05). MAFLD was not associated with TAA or aortic dissection. | MAFLD was significantly associated with AAA, which has important clinical implications. |
| Jamalinia M, 2025 [20] | Iran | In a retrospective longitudinal study of 141 consecutive AAA open repair surgery patients (92% male, mean age of 70 years (SD: 11.5)) from October 2016 to September 2021, with a median follow-up of 35 months (IQR: 0.7–56.6), the primary outcome being all-cause mortality, aHRs were calculated for each Fib-4 cut-off between 1.5 and 3.25. | FIB-4 ≥ 2.67 increased mortality by 78% (aHR:1.78, 95% CI: 1.06–3.00). Furthermore, FIB-4 ≥ 2.67 was significantly associated with a baseline aneurysm size ≥ 8cm (aOR: 2.67, 95% CI: 1.17–6.09). FIB-4 was independently associated with higher mortality risk and higher aneurysm size. | Non-invasive assessment of liver fibrosis with FIB-4 may aid in more precise risk stratification and management strategies for AAA patients in clinical practice. |
| Wang X, 2025 [14] | China | A retrospective analysis of 151 AAA subjects (mean age 69.1 ± 10.5 years) and age- and sex-matched healthy subjects who underwent abdominal CTA and non-enhanced CT scanning from January 2015 to January 2023. AAA subjects were categorized into progression (growth rate > 10 mL/year) and non-progression groups based on growth rate, as well as those with and without NAFLD, based on abdominal CT results. The Kaplan–Meier and Cox regression were used to investigate the association between NAFLD and AAA progression. | A total of 66 out of 151 AAA patients had NAFLD. Over a median of 10.7 months (6.0–76.0 months), 57 patients (37.7%) had AAA progression. The prevalence of NAFLD was significantly higher in the AAA group compared to the control group (43.7% vs. 31.1%, p = 0.024). MRA showed NAFLD to be an independent predictor of AAA progression (HR, 4.28; 95% CI, 2.20–8.31; p < 0.001). The AUC of combined NAFLD and AAA maximal diameter was 0.857 for predicting AAA progression. | NAFLD assessed with non-enhanced CT independently predicts AAA progression and can help improve the prediction of AAA progression. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jamalinia, M.; Lonardo, A.; Weiskirchen, R. Abdominal Aortic Aneurysm and Liver Fibrosis: Clinical Evidence and Molecular Pathomechanisms. Int. J. Mol. Sci. 2025, 26, 3440. https://doi.org/10.3390/ijms26073440
Jamalinia M, Lonardo A, Weiskirchen R. Abdominal Aortic Aneurysm and Liver Fibrosis: Clinical Evidence and Molecular Pathomechanisms. International Journal of Molecular Sciences. 2025; 26(7):3440. https://doi.org/10.3390/ijms26073440
Chicago/Turabian StyleJamalinia, Mohamad, Amedeo Lonardo, and Ralf Weiskirchen. 2025. "Abdominal Aortic Aneurysm and Liver Fibrosis: Clinical Evidence and Molecular Pathomechanisms" International Journal of Molecular Sciences 26, no. 7: 3440. https://doi.org/10.3390/ijms26073440
APA StyleJamalinia, M., Lonardo, A., & Weiskirchen, R. (2025). Abdominal Aortic Aneurysm and Liver Fibrosis: Clinical Evidence and Molecular Pathomechanisms. International Journal of Molecular Sciences, 26(7), 3440. https://doi.org/10.3390/ijms26073440