
Recent Update on siRNA Therapeutics

 , , ,
, , ,  and
and
Abstract
1. Introduction
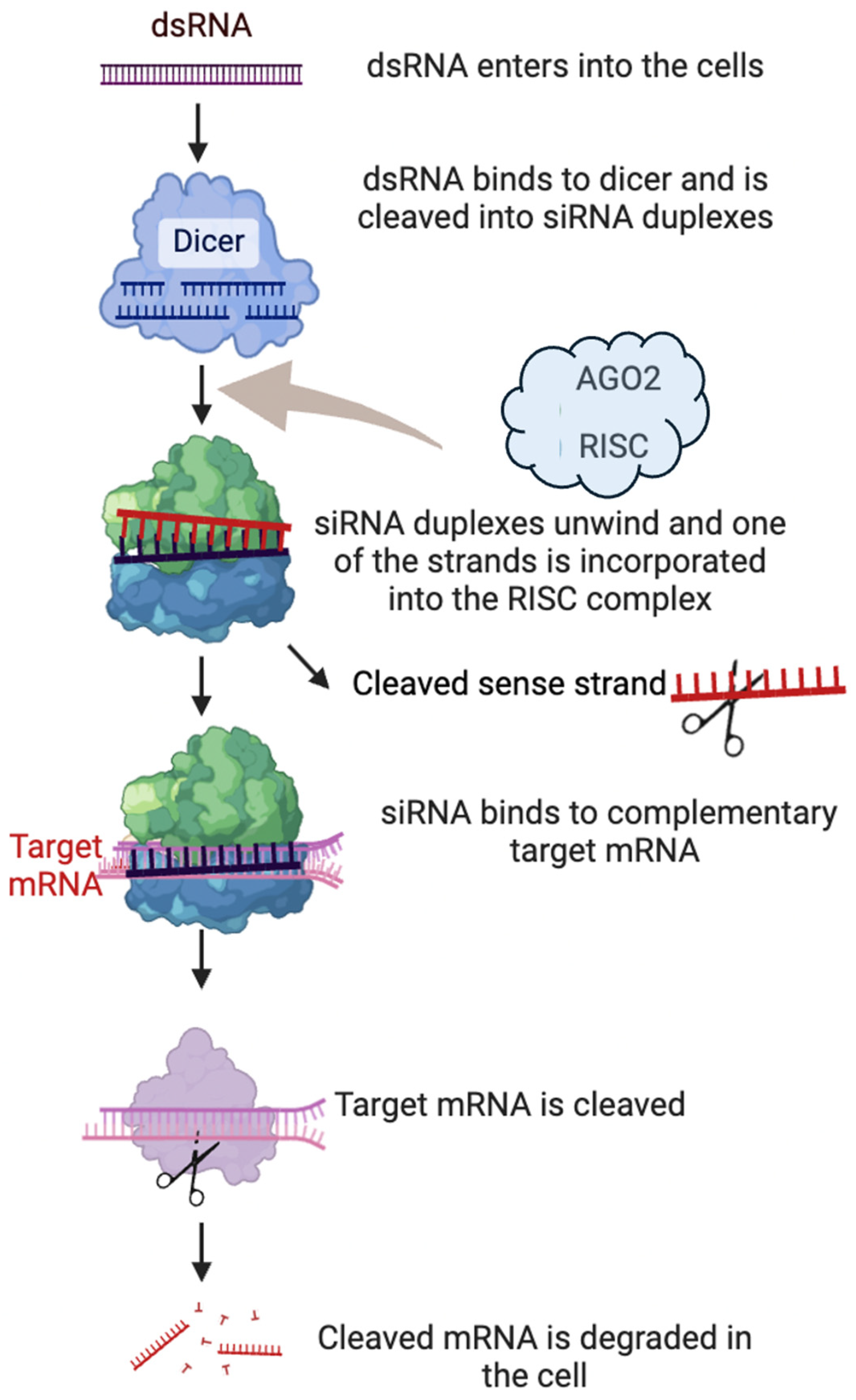
2. Mechanism of Action of RNA Interference
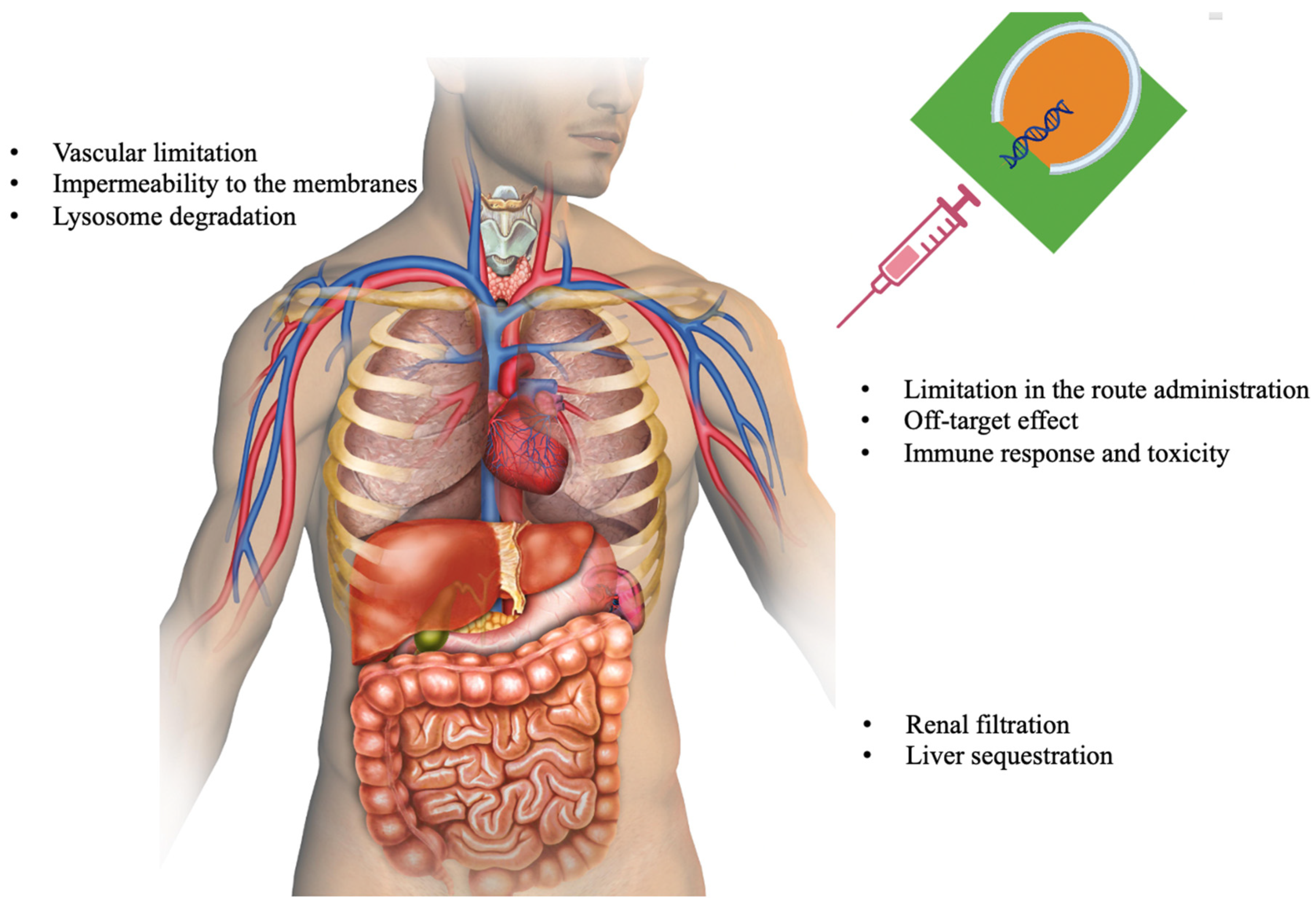
3. Challenges and the Way out
3.1. siRNA Delivery
3.2. Off-Target Effects
3.3. Bio-Stability
3.4. Immunogenicity
3.5. Limited Tissue Penetration
3.6. DNA and RNA Modification
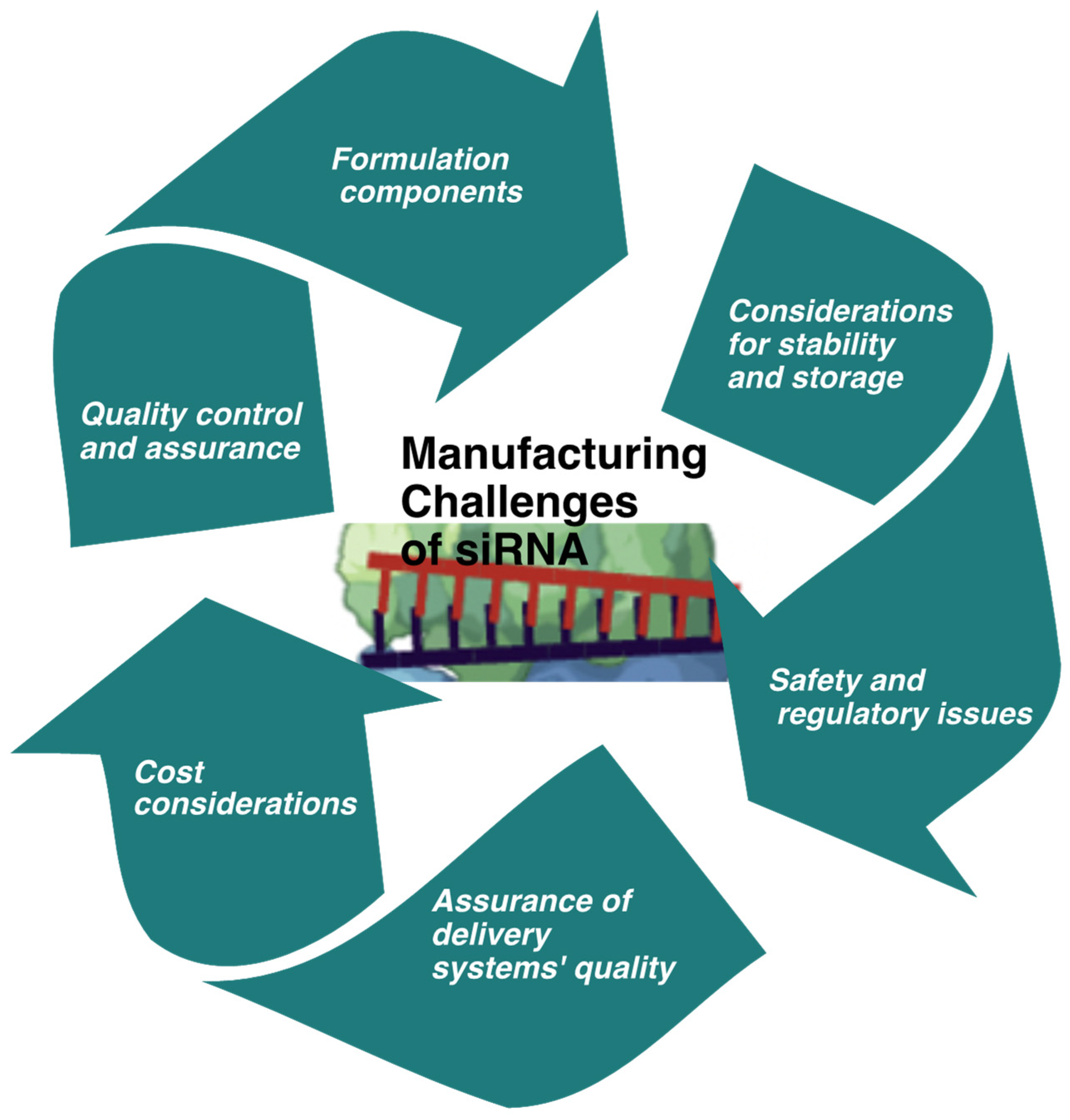
4. Manufacturing Challenges of siRNA
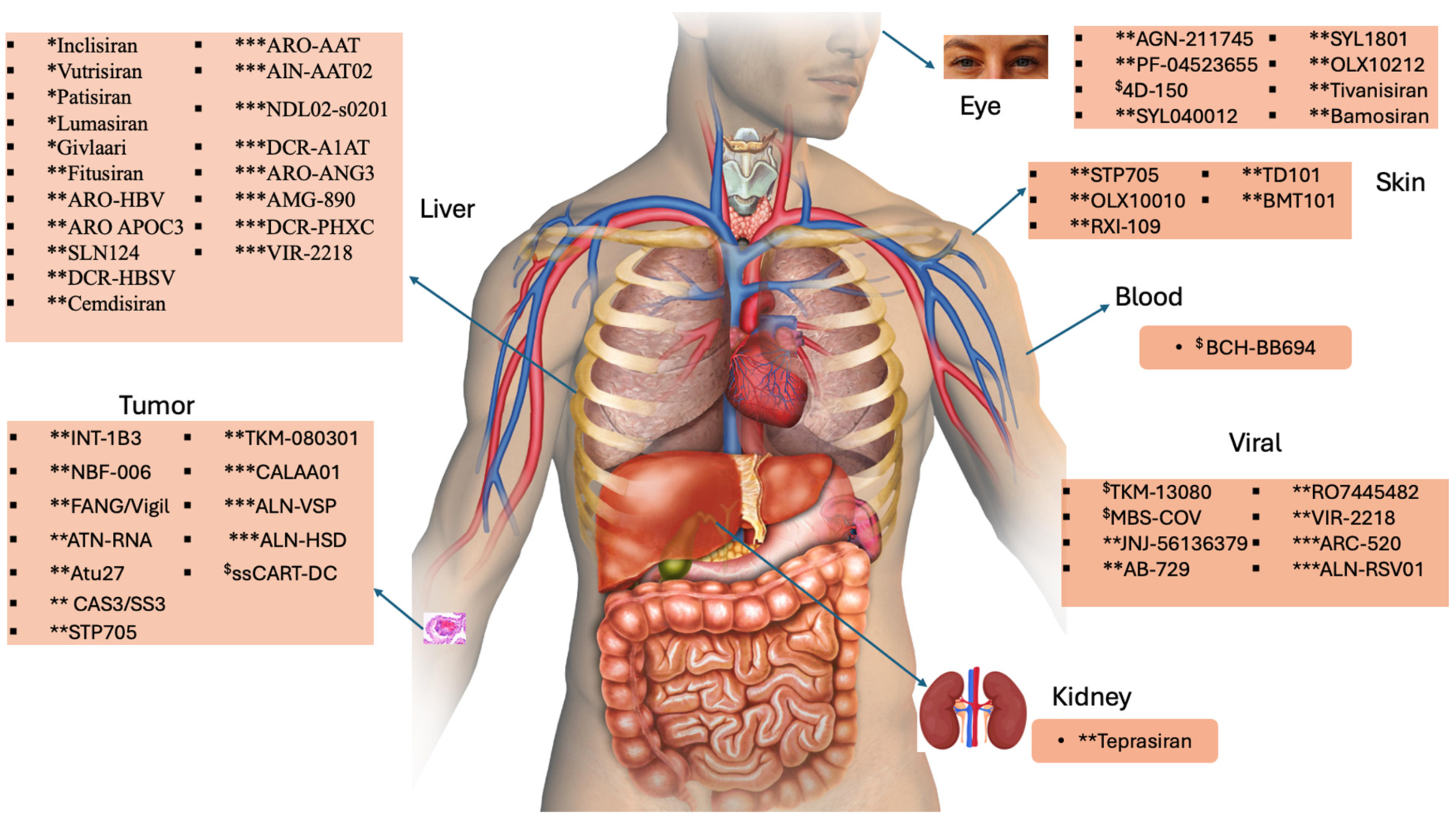
5. Naked, LNPs, and GalNAc–siRNA Conjugate Therapeutic Agents
5.1. Naked siRNA-Based Therapeutics
5.1.1. QPI-1002
5.1.2. SYL040012
5.1.3. QPI-1007
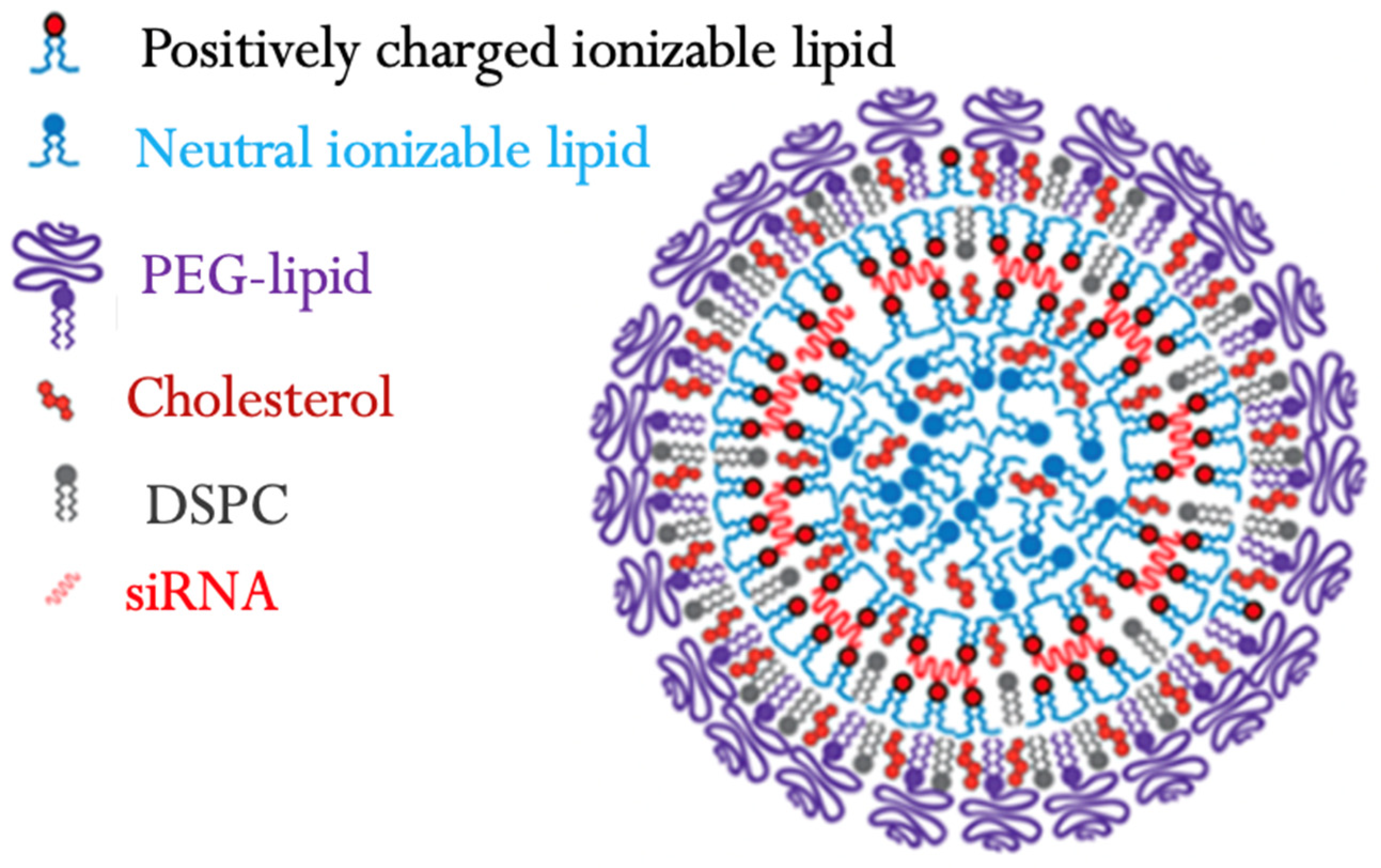
5.2. Lipid-Based siRNA
ALN-TTR02
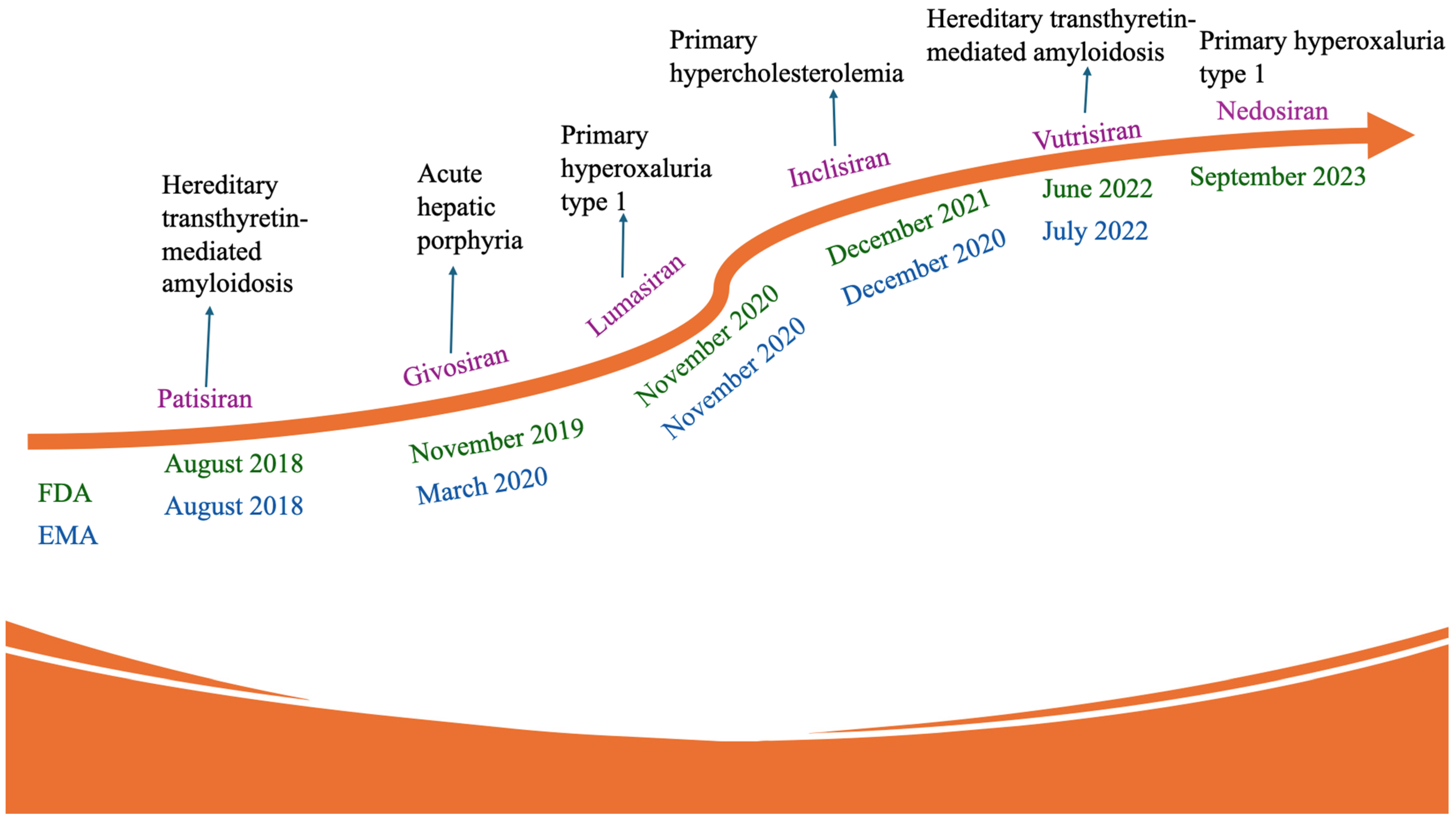
5.3. GalNAc-siRNA Conjugates
5.3.1. Givosiran
5.3.2. Vutrisiran
5.3.3. Cemdisiran
5.3.4. Nedosiran
5.3.5. Lumasiran
5.3.6. ARO-APOC3
5.3.7. SLN360
5.3.8. RBD1016
5.3.9. DCR-A1AT
5.3.10. Olpasiran
5.3.11. Inclisiran
6. Successes, Failures, and Lessons Learned So Far from the Development of siRNAs
- Broad-spectrum activity
- Potent antiviral activity
- Rapid therapeutic response
- Isoform-specific modulation
- Combinatorial Approaches
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Huang, Z.; Pei, H.; Jia, Z.; Zheng, J. Molecular glue-mediated targeted protein degradation: A novel strategy in small-molecule drug development. iScience 2024, 27, 110712. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef]
- Schubert, S.; Grünweller, A.; Erdmann, V.A.; Kurreck, J. Local RNA Target Structure Influences siRNA Efficacy: Systematic Analysis of Intentionally Designed Binding Regions. J. Mol. Biol. 2005, 348, 883–893. [Google Scholar] [CrossRef]
- Guo, P.; Coban, O.; Snead, N.M.; Trebley, J.; Hoeprich, S.; Guo, S.; Shu, Y. Engineering RNA for Targeted siRNA Delivery and Medical Application. Adv. Drug Deliv. Rev. 2010, 62, 650–666. [Google Scholar] [CrossRef]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef]
- Subhan, M.A.; Torchilin, V.P. siRNA based drug design, quality, delivery and clinical translation. Nanomedicine 2020, 29, 102239. [Google Scholar] [CrossRef]
- Shyu, A.-B.; Wilkinson, M.F.; van Hoof, A. Messenger RNA regulation: To translate or to degrade. EMBO J. 2008, 27, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Prévost, K.; Desnoyers, G.; Jacques, J.-F.; Lavoie, F.; Massé, E. Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev. 2011, 25, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.-M.; Choi, Y.H.; Tu, M.-J. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar]
- Ameres, S.L.; Martinez, J.; Schroeder, R. Molecular Basis for Target RNA Recognition and Cleavage by Human RISC. Cell 2007, 130, 101–112. [Google Scholar] [CrossRef]
- Draz, M.S.; Fang, B.A.; Zhang, P.; Hu, Z.; Gu, S.; Weng, K.C.; Gray, J.W.; Chen, F.F. Nanoparticle-Mediated Systemic Delivery of siRNA for Treatment of Cancers and Viral Infections. Theranostics 2014, 4, 872–892. [Google Scholar] [CrossRef]
- Tambuyzer, E.; Vandendriessche, B.; Austin, C.P.; Brooks, P.J.; Larsson, K.; Needleman, K.I.M.; Valentine, J.; Davies, K.; Groft, S.C.; Preti, R.; et al. Therapies for rare diseases: Therapeutic modalities, progress and challenges ahead. Nat. Rev. Drug Discov. 2020, 19, 93–111. [Google Scholar] [CrossRef]
- Lewis, D.L.; Wolff, J.A. Delivery of siRNA and siRNA expression constructs to adult mammals by hydrodynamic intravascular injection. Methods Enzymol. 2005, 392, 336–350. [Google Scholar]
- van de Water, F.M.; Boerman, O.C.; Wouterse, A.C.; Peters, J.G.P.; Russel, F.G.M.; Masereeuw, R. Intravenously administered short interfering rna accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. [Google Scholar] [CrossRef]
- Dorsett, Y.; Tuschl, T. siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. 2017, 13, 48–57. [Google Scholar]
- Alshaer, W.; Zureigat, H.; Al Karaki, A.; Al-Kadash, A.; Gharaibeh, L.; Hatmal, M.M.; Aljabali, A.A.; Awidi, A. siRNA: Mechanism of action, challenges, and therapeutic approaches. Eur. J. Pharmacol. 2021, 905, 174178. [Google Scholar] [CrossRef] [PubMed]
- Merdan, T.; Kopeček, J.; Kissel, T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv. Drug Deliv. Rev. 2002, 54, 715–758. [Google Scholar] [PubMed]
- Patil, Y.; Panyam, J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int. J. Pharm. 2009, 367, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Physicochemical and Biological Characterization of Targeted, Nucleic Acid-Containing Nanoparticles. Bioconj. Chem. 2007, 18, 456–468. [Google Scholar] [CrossRef]
- Kumar, P.; Ban, H.-S.; Kim, S.-S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific siRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar]
- Liu, F.; Song, Y.K.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef]
- Stoll, S.M.; Sclimenti, C.R.; Baba, E.J.; Meuse, L.; Kay, M.A.; Calos, M.P. Epstein-Barr virus/human vector provides high-level, long-term expression of alpha1-antitrypsin in mice. Mol. Ther. 2001, 4, 122–129. [Google Scholar] [CrossRef]
- Tseng, Y.-C.; Mozumdar, S.; Huang, L. Lipid-based systemic delivery of siRNA. Adv. Drug Deliv. Rev. 2009, 61, 721–731. [Google Scholar]
- Durcan, N.; Murphy, C.; Cryan, S.-A. Inhalable siRNA: Potential as a Therapeutic Agent in the Lungs. Mol. Pharm. 2008, 5, 559–566. [Google Scholar] [PubMed]
- Vicentini, F.T.M.d.C.; Borgheti-Cardoso, L.N.; Depieri, L.V.; Mano, D.d.M.; Abelha, T.F.; Petrilli, R.; Bentley, M.V.L.B. Delivery Systems and Local Administration Routes for Therapeutic siRNA. Pharm. Res. 2013, 30, 915–931. [Google Scholar] [PubMed]
- Debacker, A.J.; Voutila, J.; Catley, M.; Blakey, D.; Habib, N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol. Ther. 2020, 28, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.A.; Nam, K.T.; Lee, K.; et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.; Wang, Y.; Kong, J.; Pan, T.; Yang, Y.; He, J. Lipid nanoparticles as the drug carrier for targeted therapy of hepatic disorders. J. Mater. Chem. B 2024, 12, 4759–4784. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Ricci, E.P.; Mercier, B.C.; Moore, M.J.; Fitzgerald, K.A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 2014, 14, 361–376. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Khan, A.A.; Betel, D.; Miller, M.L.; Sander, C.; Leslie, C.S.; Marks, D.S. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 2009, 27, 549–555. [Google Scholar] [CrossRef]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Ding, H.; Kennington, L.; Moore, J.T.; Schelter, J.; Burchard, J.; Linsley, P.S.; Aronin, N.; Xu, Z.; Zamore, P.D. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet 2006, 2, e140. [Google Scholar] [CrossRef]
- Sioud, M. Does the understanding of immune activation by RNA predict the design of safe siRNAs? Front. Biosci. 2008, 13, 4379–4392. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Blanchard, K.; Shaw, L.; Jensen, K.; Lockridge, J.A.; Dickinson, B.; McSwiggen, J.A.; Vargeese, C.; Bowman, K.; Shaffer, C.S.; et al. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 2005, 41, 1349–1356. [Google Scholar] [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [PubMed]
- de Fougerolles, A.; Vornlocher, H.-P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar]
- Elmen, J.; Thonberg, H.; Ljungberg, K.; Frieden, M.; Westergaard, M.; Xu, Y.; Wahren, B.; Liang, Z.; Orum, H.; Koch, T.; et al. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005, 33, 439–447. [Google Scholar]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar]
- Zheng, Y.Y.; Wu, Y.; Begley, T.J.; Sheng, J. Sulfur modification in natural RNA and therapeutic oligonucleotides. RSC Chem. Biol. 2021, 2, 990–1003. [Google Scholar] [CrossRef]
- Larsen, A.T.; Fahrenbach, A.C.; Sheng, J.; Pian, J.; Szostak, J.W. Thermodynamic insights into 2-thiouridine-enhanced RNA hybridization. Nucleic Acids Res. 2015, 43, 7675–7687. [Google Scholar]
- Dimitrova, D.G.; Teysset, L.; Carré, C. RNA 2′-O-Methylation (Nm) Modification in Human Diseases. Genes 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Kushwah, V.; Jain, S. Recent advances in tumor targeting approaches. In Targeted Drug Delivery: Concepts and Design; Springer: Cham, Switzerland, 2015; pp. 41–112. [Google Scholar]
- Miele, E.; Spinelli, G.P.; Miele, E.; Di Fabrizio, E.; Ferretti, E.; Tomao, S.; Gulino, A. Nanoparticle-based delivery of small interfering RNA: Challenges for cancer therapy. Int. J. Nanomed. 2012, 7, 3637–3657. [Google Scholar]
- Selvam, C.; Mutisya, D.; Prakash, S.; Ranganna, K.; Thilagavathi, R. Therapeutic potential of chemically modified siRNA: Recent trends. Chem. Biol. Drug Des. 2017, 90, 665–678. [Google Scholar] [CrossRef]
- Iribe, H.; Miyamoto, K.; Takahashi, T.; Kobayashi, Y.; Leo, J.; Aida, M.; Ui-Tei, K. Chemical Modification of the siRNA Seed Region Suppresses Off-Target Effects by Steric Hindrance to Base-Pairing with Targets. ACS Omega 2017, 2, 2055–2064. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Liu, Y.; Yang, G.; Falconer, R.J.; Zhao, C.-X. Lipid nanoparticles for drug delivery. Adv. Nanobiomed. Res. 2022, 2, 2100109. [Google Scholar] [CrossRef]
- Qiu, M.; Tang, Y.; Chen, J.; Muriph, R.; Ye, Z.; Huang, C.; Evans, J.; Henske, E.P.; Xu, Q. Lung-selective mRNA delivery of synthetic lipid nanoparticles for the treatment of pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2116271119. [Google Scholar] [CrossRef] [PubMed]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010, 385, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Pan, W.; Su, T.; Zhang, M.; Dong, W.; Qi, X. Recent advances in natural polymer-based drug delivery systems. React. Funct. Polym. 2020, 148, 104501. [Google Scholar] [CrossRef]
- Schaffert, D.; Wagner, E. Gene therapy progress and prospects: Synthetic polymer-based systems. Gene Ther. 2008, 15, 1131–1138. [Google Scholar] [CrossRef]
- Lorenzer, C.; Dirin, M.; Winkler, A.-M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef]
- Chen, Y.; Gu, H.; Zhang, D.S.-Z.; Li, F.; Liu, T.; Xia, W. Highly effective inhibition of lung cancer growth and metastasis by systemic delivery of siRNA via multimodal mesoporous silica-based nanocarrier. Biomaterials 2014, 35, 10058–10069. [Google Scholar]
- Ahn, I.; Kang, C.S.; Han, J. Where should siRNAs go: Applicable organs for siRNA drugs. Exp. Mol. Med. 2023, 55, 1283–1292. [Google Scholar] [PubMed]
- Yu, B.; Tai, H.C.; Xue, W.; Lee, L.J.; Lee, R.J. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol. Membr. Biol. 2010, 27, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Zhi, D.; Yang, T.; Yang, J.; Fu, S.; Zhang, S. Targeting strategies for superparamagnetic iron oxide nanoparticles in cancer therapy. Acta Biomater. 2020, 102, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Luft, C.; Ketteler, R. Electroporation Knows No Boundaries: The Use of Electrostimulation for siRNA Delivery in Cells and Tissues. J. Biomol. Screen. 2015, 20, 932–942. [Google Scholar] [CrossRef]
- Yarmush, M.L.; Golberg, A.; Serša, G.; Kotnik, T.; Miklavčič, D. Electroporation-Based Technologies for Medicine: Principles, Applications, and Challenges. Annu. Rev. Biomed. Eng. 2014, 16, 295–320. [Google Scholar]
- Ovcharenko, D.; Jarvis, R.; Hunicke-Smith, S.; Kelnar, K.; Brown, D. High-throughput RNAi screening in vitro: From cell lines to primary cells. RNA 2005, 11, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Phenix, C.P.; Togtema, M.; Pichardo, S.; Zehbe, I.; Curiel, L. High Intensity Focused Ultrasound Technology, its Scope and Applications in Therapy and Drug Delivery. J. Pharm. Pharm. Sci. 2014, 17, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Focused ultrasound-mediated drug delivery through the blood–brain barrier. Expert Rev. Neurother. 2015, 15, 477–491. [Google Scholar] [CrossRef]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef]
- Parashar, D.; Rajendran, V.; Shukla, R.; Sistla, R. Lipid-based nanocarriers for delivery of small interfering RNA for therapeutic use. Eur. J. Pharm. Sci. 2020, 142, 105159. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Bi, D.; Plantinga, J.A.; Molema, G.; Bussmann, J.; Kamps, J.A.A.M. Development of a Combined Lipid-Based Nanoparticle Formulation for Enhanced siRNA Delivery to Vascular Endothelial Cells. Pharmaceutics 2022, 14, 2086. [Google Scholar] [CrossRef]
- Anwar, S.; Mir, F.; Yokota, T. Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies. Pharmaceutics 2023, 15, 1130. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Xu, H.; Liao, C.; Liang, S.; Ye, B.-C. A Novel Peptide-Equipped Exosomes Platform for Delivery of Antisense Oligonucleotides. ACS Appl. Mater. Interfaces 2021, 13, 10760–10767. [Google Scholar] [CrossRef]
- Jin, Y.; Lee, J.S.; Min, S.; Park, H.; Kang, T.J.; Cho, S. Bioengineered Extracellular Membranous Nanovesicles for Efficient Small-Interfering RNA Delivery: Versatile Platforms for Stem Cell Engineering and In Vivo Delivery. Adv. Funct. Mater. 2016, 26, 5804–5817. [Google Scholar] [CrossRef]
- Wang, M.-K.; Gao, C.-C.; Yang, Y.-G. Emerging Roles of RNA Methylation in Development. Acc. Chem. Res. 2023, 56, 3417–3427. [Google Scholar] [CrossRef]
- Zhu, D.-H.; Su, K.-K.; Ou-Yang, X.-X.; Zhang, Y.-H.; Yu, X.-P.; Li, Z.-H.; Ahmadi-Nishaboori, S.-S.; Li, L.-J. Mechanisms and clinical landscape of N6-methyladenosine (m6A) RNA modification in gastrointestinal tract cancers. Mol. Cell. Biochem. 2024, 479, 1553–1570. [Google Scholar] [PubMed]
- Lee, S.H.; Kang, Y.Y.; Jang, H.-E.; Mok, H. Current preclinical small interfering RNA (siRNA)-based conjugate systems for RNA therapeutics. Adv. Drug Deliv. Rev. 2016, 104, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Lyko, F.; Helm, M. 5-methylcytosine in RNA: Detection, enzymatic formation and biological functions. Nucleic Acids Res. 2010, 38, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.; Rehman, M.U.; Tayara, H.; Lee, S.W.; Chong, K.T. m5C-Seq: Machine learning-enhanced profiling of RNA 5-methylcytosine modifications. Comput. Biol. Med. 2024, 182, 109087. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.; Rehman, M.U.; Tayara, H.; Zou, Q.; Chong, K.T. XGBoost framework with feature selection for the prediction of RNA N5-methylcytosine sites. Mol. Ther. 2023, 31, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-H.; Guo, T.; Wang, M.; Liu, J.-H.; Zheng, L.-M.; He, Y. RNA m6A modification facilitates DNA methylation during maize kernel development. Plant Physiol. 2024, 194, 2165–2182. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Shen, J.-Y.; Chen, D.-P.; Wu, C.-F.; Guo, R.; Zhang, P.-P.; Lv, J.-W.; Li, W.-F.; Wang, Z.-X. Identification of cross-talk between m6A and 5mC regulators associated with onco-immunogenic features and prognosis across 33 cancer types. J. Hematol. Oncol. 2020, 13, 22. [Google Scholar] [CrossRef]
- Wang, R.; Chung, C.-R.; Lee, T.-Y. Interpretable Multi-Scale Deep Learning for RNA Methylation Analysis across Multiple Species. Int. J. Mol. Sci. 2024, 25, 2869. [Google Scholar] [CrossRef]
- Abbas, Z.; Tayara, H.; Chong, K.T. ZayyuNet—A Unified Deep Learning Model for the Identification of Epigenetic Modifications Using Raw Genomic Sequences. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 2533–2544. [Google Scholar]
- Beaucage, S.L. Solid-phase synthesis of siRNA oligonucleotides. Curr. Opin. Drug Discov. Dev. 2008, 11, 203–216. [Google Scholar]
- Schaffert, D.; Troiber, C.; Salcher, E.E.; Fröhlich, T.; Martin, I.; Badgujar, N.; Dohmen, C.; Edinger, D.; Kläger, R.; Maiwald, G.; et al. Solid-phase synthesis of sequence-defined T-, i-, and U-shape polymers for pDNA and siRNA delivery. Angew. Chem. Int. Ed. Engl. 2011, 50, 8986–8989. [Google Scholar]
- Lönnberg, H. Solid-Phase Synthesis of Oligonucleotide Conjugates Useful for Delivery and Targeting of Potential Nucleic Acid Therapeutics. Bioconj. Chem. 2009, 20, 1065–1094. [Google Scholar] [CrossRef]
- Amarzguioui, M.; Rossi, J.J.; Kim, D. Approaches for chemically synthesized siRNA and vector-mediated RNAi. FEBS Lett. 2005, 579, 5974–5981. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, I.; Eells, K.; Hudson, I. A Comparison of Currently Approved Small Interfering RNA (siRNA) Medications to Alternative Treatments by Costs, Indications, and Medicaid Coverage. Pharmacy 2024, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.cda-amc.ca/sites/default/files/DRR/2021/SR0679%20Givlaari%20-%20CADTH%20Final%20Rec-pw.pdf (accessed on 20 February 2023).
- Available online: https://www.cda-amc.ca/sites/default/files/DRR/2023/SR0734%20Oxlumo%20-%20Final%20CADTH%20Recommendation%20(with%20redactions)%20Final.pdf (accessed on 20 February 2025).
- Available online: https://www.ncbi.nlm.nih.gov/books/NBK602494/table/t05/?report=objectonly (accessed on 20 February 2025).
- Available online: https://www.novocare.com/eligibility/rivfloza-offer.html (accessed on 20 February 2025).
- O’mahony, A.M.; Godinho, B.M.; Cryan, J.F.; O’driscoll, C.M. Non-Viral Nanosystems for Gene and Small Interfering RNA Delivery to the Central Nervous System: Formulating the Solution. J. Pharm. Sci. 2013, 102, 3469–3484. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Cho, K.Y.; Tiwari, R.K. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Sun, T.; Ferrari, M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012, 19, 367–373. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Xu, C.-F.; Wang, J. Delivery systems for siRNA drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Haussecker, D. Current issues of RNAi therapeutics delivery and development. J. Control. Release 2014, 195, 49–54. [Google Scholar] [CrossRef]
- Khalil, I.A.; Yamada, Y.; Harashima, H. Optimization of siRNA delivery to target sites: Issues and future directions. Expert Opin. Drug Deliv. 2018, 15, 1053–1065. [Google Scholar] [CrossRef]
- Oh, Y.-K.; Park, T.G. siRNA delivery systems for cancer treatment. Adv. Drug Deliv. Rev. 2009, 61, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Kandil, R.; Xie, Y.; Heermann, R.; Isert, L.; Jung, K.; Mehta, A.; Merkel, O.M. Coming in and Finding Out: Blending Receptor-Targeted Delivery and Efficient Endosomal Escape in a Novel Bio-Responsive siRNA Delivery System for Gene Knockdown in Pulmonary T Cells. Adv. Ther. 2019, 2, 1900047. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.H.; Schuh, R.S.; Michels, L.R.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Lenz, G.S.; de Oliveira, F.H.; Venturin, G.; Greggio, S.; et al. Nasal Administration of Cationic Nanoemulsions as CD73-siRNA Delivery System for Glioblastoma Treatment: A New Therapeutical Approach. Mol. Neurobiol. 2020, 57, 635–649. [Google Scholar] [CrossRef]
- Alameh, M.; Lavertu, M.; Tran-Khanh, N.; Chang, C.-Y.; Lesage, F.; Bail, M.; Darras, V.; Chevrier, A.; Buschmann, M.D. siRNA Delivery with Chitosan: Influence of Chitosan Molecular Weight, Degree of Deacetylation, and Amine to Phosphate Ratio on in Vitro Silencing Efficiency, Hemocompatibility, Biodistribution, and in Vivo Efficacy. Biomacromolecules 2018, 19, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [PubMed]
- Leung, A.K.K.; Tam, Y.Y.C.; Cullis, P.R. Chapter Four—Lipid Nanoparticles for Short Interfering RNA Delivery. In Advances in Genetics; Huang, L., Liu, D., Wagner, E., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 71–110. [Google Scholar]
- Chen, C.; Yang, Z.; Tang, X. Chemical modifications of nucleic acid drugs and their delivery systems for gene-based therapy. Med. Res. Rev. 2018, 38, 829–869. [Google Scholar]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar]
- Willoughby, J.L.; Chan, A.; Sehgal, A.; Butler, J.S.; Nair, J.K.; Racie, T.; Shulga-Morskaya, S.; Nguyen, T.; Qian, K.; Yucius, K.; et al. Evaluation of GalNAc-siRNA Conjugate Activity in Pre-clinical Animal Models with Reduced Asialoglycoprotein Receptor Expression. Mol. Ther. 2018, 26, 105–114. [Google Scholar] [CrossRef]
- Rajeev, K.G.; Nair, J.K.; Jayaraman, M.; Charisse, K.; Taneja, N.; O’Shea, J.; Willoughby, J.L.S.; Yucius, K.; Nguyen, T.; Shulga-Morskaya, S.; et al. Hepatocyte-Specific Delivery of siRNAs Conjugated to Novel Non-nucleosidic Trivalent N-Acetylgalactosamine Elicits Robust Gene Silencing in Vivo. ChemBioChem 2015, 16, 903–908. [Google Scholar]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef]
- Korin, E.; Bejerano, T.; Cohen, S. GalNAc bio-functionalization of nanoparticles assembled by electrostatic interactions improves siRNA targeting to the liver. J. Control. Release 2017, 266, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Kubczak, M.; Michlewska, S.; Bryszewska, M.; Aigner, A.; Ionov, M. Nanoparticles for local delivery of siRNA in lung therapy. Adv. Drug Deliv. Rev. 2021, 179, 114038. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mangala, L.S.; Rodriguez-Aguayo, C.; Kong, X.; Lopez-Berestein, G.; Sood, A.K. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev. 2018, 37, 107–124. [Google Scholar] [CrossRef]
- Aigner, A. Delivery Systems for the Direct Application of siRNAs to Induce RNA Interference (RNAi) In Vivo. BioMed Res. Int. 2006, 2006, 71659. [Google Scholar] [CrossRef]
- Zaidi, S.S.A.; Fatima, F.; Zhou, D.; Deng, W.; Liu, S. Engineering siRNA therapeutics: Challenges and strategies. J. Nanobiotechnol. 2023, 21, 381. [Google Scholar]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, I.K.; Bae, K.H.; Lee, S.H.; Lee, Y.; Park, T.G. Cationic Solid Lipid Nanoparticles Reconstituted from Low Density Lipoprotein Components for Delivery of siRNA. Mol. Pharm. 2008, 5, 622–631. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef]
- Gangopadhyay, S.; Gore, K.R. Advances in siRNA therapeutics and synergistic effect on siRNA activity using emerging dual ribose modifications. RNA Biol. 2022, 19, 452–467. [Google Scholar] [PubMed]
- Demirjian, S.; Ailawadi, G.; Polinsky, M.; Bitran, D.; Silberman, S.; Shernan, S.K.; Burnier, M.; Hamilton, M.; Squiers, E.; Erlich, S.; et al. Safety and Tolerability Study of an Intravenously Administered Small Interfering Ribonucleic Acid (siRNA) Post On-Pump Cardiothoracic Surgery in Patients at Risk of Acute Kidney Injury. Kidney Int. Rep. 2017, 2, 836–843. [Google Scholar] [PubMed]
- Thielmann, M.; Corteville, D.; Szabo, G.; Swaminathan, M.; Lamy, A.; Lehner, L.J.; Brown, C.D.; Noiseux, N.; Atta, M.G.; Squiers, E.C.; et al. Teprasiran, a Small Interfering RNA, for the Prevention of Acute Kidney Injury in High-Risk Patients Undergoing Cardiac Surgery: A Randomized Clinical Study. Circulation 2021, 144, 1133–1144. [Google Scholar] [PubMed]
- Martínez, T.; González, M.V.; Roehl, I.; Wright, N.; Pañeda, C.; Jiménez, A.I. In Vitro and In Vivo Efficacy of SYL040012, a Novel siRNA Compound for Treatment of Glaucoma. Mol. Ther. 2014, 22, 81–91. [Google Scholar] [CrossRef]
- Moreno-Montañés, J.; Sádaba, B.; Ruz, V.; Gómez-Guiu, A.; Zarranz, J.; González, M.V.; Pañeda, C.; Jimenez, A.I. Phase I clinical trial of SYL040012, a small interfering RNA targeting β-adrenergic receptor 2, for lowering intraocular pressure. Mol. Ther. 2014, 22, 226–232. [Google Scholar]
- Gonzalez, V.; Palumaa, K.; Turman, K.; Muñoz, F.J.; Jordan, J.; García, J.; Ussa, F.; Antón, A.; Gutierrez, E.; Moreno-Montanes, J. Phase 2 of bamosiran (SYL040012), a novel RNAi based compound for the treatment of increased intraocular pressure associated to glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 564. [Google Scholar]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Solano, E.C.; Kornbrust, D.J.; Beaudry, A.; Foy, J.W.; Schneider, D.J.; Thompson, J.D. Toxicological and pharmacokinetic properties of QPI-1007, a chemically modified synthetic siRNA targeting caspase 2 mRNA, following intravitreal injection. Nucleic Acid Ther. 2014, 24, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Kalinski, H.; Berry, M.; Almasieh, M.; Ashush, H.; Slager, N.; Brafman, A.; Spivak, I.; Prasad, N.; Mett, I.; et al. Ocular neuroprotection by siRNA targeting caspase-2. Cell Death Dis. 2011, 2, e173. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154-155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.R.; Felgner, P.L. The 60-year evolution of lipid nanoparticles for nucleic acid delivery. Nat. Rev. Drug Discov. 2024, 23, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H.; et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients with Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019, 139, 431–443. [Google Scholar] [CrossRef]
- Li, T.; Wu, M.; Zhu, Y.Y.; Chen, J.; Chen, L. Development of RNA Interference–Based Therapeutics and Application of Multi-Target Small Interfering RNAs. Nucleic Acid Ther. 2014, 24, 302–312. [Google Scholar] [CrossRef]
- An, G. Pharmacokinetics and Pharmacodynamics of GalNAc-Conjugated siRNAs. J. Clin. Pharmacol. 2024, 64, 45–57. [Google Scholar] [CrossRef]
- Available online: https://capella.alnylam.com/wp-content/uploads/2020/09/OTS-2020_MacLauchlin.pdf (accessed on 19 February 2025).
- Sardh, E.; Harper, P.; Balwani, M.; Stein, P.; Rees, D.; Bissell, D.M.; Desnick, R.; Parker, C.; Phillips, J.; Bonkovsky, H.L.; et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N. Engl. J. Med. 2019, 380, 549–558. [Google Scholar] [CrossRef]
- Sardh, E.; Balwani, M.; Harper, P.; Stein, P.; Rees, D.; Bloomer, J.; Bissell, D.; Parket, C.; Phillips, J.; Bonkovsky, H.; et al. A phase 1/2, randomized, placebo controlled and open label extension studies of Givosiran and investigational RNA interference therapeutic, in patients with acute intermittent porphyria. J. Hepatol. 2018, 68, S66–S67. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Ventura, P.; Bonkovsky, H.L.; Gouya, L.; Aguilera-Peiró, P.; Bissell, D.M.; Stein, P.E.; Balwani, M.; Anderson, D.K.E.; Parker, C.; Kuter, D.J.; et al. Efficacy and safety of givosiran for acute hepatic porphyria: 24-month interim analysis of the randomized phase 3 ENVISION study. Liver Int. 2021, 42, 161–172. [Google Scholar] [CrossRef]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382. [Google Scholar] [PubMed]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.-C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2023, 30, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Gaya, A.; Munir, T.; Urbano-Ispizua, A.; Griffin, M.; Taubel, J.; Bush, J.; Bhan, I.; Borodovsky, A.; Wang, Y.; Badri, P. Results of a phase 1/2 study of cemdisiran in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. eJHaem 2023, 4, 612–624. [Google Scholar]
- Hoppe, B.; Koch, A.; Cochat, P.; Garrelfs, S.F.; Baum, M.A.; Groothoff, J.W.; Lipkin, G.; Coenen, M.; Schalk, G.; Amrite, A.; et al. Safety, pharmacodynamics, and exposure-response modeling results from a first-in-human phase 1 study of nedosiran (PHYOX1) in primary hyperoxaluria. Kidney Int. 2022, 101, 626–634. [Google Scholar] [CrossRef]
- Ariceta, G.; Barrios, K.; Brown, B.D.; Hoppe, B.; Rosskamp, R.; Langman, C.B. Hepatic Lactate Dehydrogenase A: An RNA Interference Target for the Treatment of All Known Types of Primary Hyperoxaluria. Kidney Int. Rep. 2021, 6, 1088–1098. [Google Scholar] [CrossRef]
- Baum, M.A.; Langman, C.; Cochat, P.; Lieske, J.C.; Moochhala, S.H.; Hamamoto, S.; Satoh, H.; Mourani, C.; Ariceta, G.; Torres, A.; et al. PHYOX2: A pivotal randomized study of nedosiran in primary hyperoxaluria type 1 or 2. Kidney Int. 2023, 103, 207–217. [Google Scholar]
- Available online: https://classic.clinicaltrials.gov/ct2/results?cond=&term=dCR-PHXC&cntry=&state=&city=&dist= (accessed on 19 February 2025).
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; Hoff, W.G.V.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef]
- Hayes, W.; Sas, D.J.; Magen, D.; Shasha-Lavsky, H.; Michael, M.; Sellier-Leclerc, A.-L.; Hogan, J.; Ngo, T.; Sweetser, M.T.; Gansner, J.M.; et al. Efficacy and safety of lumasiran for infants and young children with primary hyperoxaluria type 1: 12-month analysis of the phase 3 ILLUMINATE-B trial. Pediatr. Nephrol. 2023, 38, 1075–1086. [Google Scholar] [CrossRef]
- Michael, M.; Groothoff, J.W.; Shasha-Lavsky, H.; Lieske, J.C.; Frishberg, Y.; Simkova, E.; Sellier-Leclerc, A.-L.; Devresse, A.; Guebre-Egziabher, F.; Bakkaloglu, S.A.; et al. Lumasiran for Advanced Primary Hyperoxaluria Type 1: Phase 3 ILLUMINATE-C Trial. Am. J. Kidney Dis. 2023, 81, 145–155.e1. [Google Scholar] [CrossRef]
- Wong, S.C.; Li, Z.; Given, B.; Seefeld, M.; Andersen, A.; Zhu, R.; Havel, P.; Hamilton, J.; Graham, J.; Pei, T.; et al. Personalized Medicine for Dyslipidemias by RNA Interference-Mediated Reductions in Apolipoprotein C3 or Angiopoietin-Like Protein 3. J. Clin. Lipidol. 2019, 13, e15. [Google Scholar]
- Schwabe, C.; Scott, R.; Sullivan, D.; Baker, J.; Clifton, P.; Hamilton, J.; Given, B.; Martin, J.S.; Melquist, S.; Watts, G.; et al. RNA interference targeting apolipoprotein C-III with ARO-APOC3 in healthy volunteers mimics lipid and lipoprotein findings seen in subjects with inherited apolipoprotein C-III deficiency. Eur. Hear. J. 2020, 41 (Suppl. S2), ehaa946.3330. [Google Scholar]
- Clifton, P.; Sullivan, D.; Baker, J.; Schwabe, C.; Thackwray, S.; Scott, R.; Hamilton, J.; Chang, T.; Given, B.; Martin, J.S.; et al. Pharmacodynamic Effect of ARO-APOC3, an Investigational Hepatocyte-targeted RNA Interference Therapeutic Targeting Apolipoprotein C3, in Patients with Hypertriglyceridemia and Multifactorial Chylomicronemia. Circulation 2020, 142 (Suppl. S3), A12594. [Google Scholar]
- Clifton, P.; Sullivan, D.; Baker, J.; Schwabe, C.; Thackwray, S.; Scott, R.; Hamilton, J.; Given, B.; Martin, J.S.; Melquist, S.; et al. First Results of RNA Interference Against Apolipoprotein C3 as a Treatment for Chylomicronemia. J. Clin. Lipidol. 2020, 14, 594. [Google Scholar] [CrossRef]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar]
- Available online: https://www.ribolia.com/En/major-procucts (accessed on 20 February 2025).
- Available online: https://www.natap.org/2023/EASL/EASL_121.htm (accessed on 20 February 2025).
- Available online: https://www.businesswire.com/news/home/20211112005405/en/ (accessed on 20 February 2025).
- O’brien, M.E.; Murray, G.; Gogoi, D.; Yusuf, A.; McCarthy, C.; Wormald, M.R.; Casey, M.; Gabillard-Lefort, C.; McElvaney, N.G.; Reeves, E.P. A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application. Int. J. Mol. Sci. 2022, 23, 2441. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- Sohn, W.; Winkle, P.; Neutel, J.; Wu, Y.; Jabari, F.; Terrio, C.; Varrieur, T.; Wang, J.; Hellawell, J. Pharmacokinetics, Pharmacodynamics, and Tolerability of Olpasiran in Healthy Japanese and Non-Japanese Participants: Results from a Phase I, Single-dose, Open-label Study. Clin. Ther. 2022, 44, 1237–1247. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Hear. J. 2022, 251, 61–69. [Google Scholar]
- O’donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, O.; Comoglio, P.; Wong, G.K.-S.; Tuszynski, J.A. Development of Novel siRNA Therapeutics: A Review with a Focus on Inclisiran for the Treatment of Hypercholesterolemia. Int. J. Mol. Sci. 2023, 24, 4019. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Nishikido, T.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.L.J.; Kastelein, J.J.P. Effect of 1 or 2 Doses of Inclisiran on Low-Density Lipoprotein Cholesterol Levels: One-Year Follow-up of the ORION-1 Randomized Clinical Trial. JAMA Cardiol. 2019, 4, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Hovingh, G.K.; Lepor, N.E.; Kallend, D.; Stoekenbroek, R.M.; Wijngaard, P.L.J.; Raal, F.J. Inclisiran Durably Lowers Low-Density Lipoprotein Cholesterol and Proprotein Convertase Subtilisin/Kexin Type 9 Expression in Homozygous Familial Hypercholesterolemia: The ORION-2 Pilot Study. Circulation 2020, 141, 1829–1831. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Wright, R.S.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef]
- Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.J.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Lawrence, D.; Friedman, A.; et al. Inclisiran and cardiovascular events: A patient-level analysis of phase III trials. Eur. Hear. J. 2022, 44, 129–138. [Google Scholar] [CrossRef]
- Kallend, D.; Stoekenbroek, R.; He, Y.; Smith, P.F.; Wijngaard, P. Pharmacokinetics and pharmacodynamics of inclisiran, a small interfering RNA therapy, in patients with hepatic impairment. J. Clin. Lipidol. 2022, 16, 208–219. [Google Scholar] [CrossRef]
- Available online: https://www.tctmd.com/news/orion-8-safe-and-durable-longer-term-ldl-lowering-inclisiran (accessed on 20 February 2025).
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ (accessed on 3 March 2025).
- Available online: https://www.asgct.org/global/documents/asgct-citeline-q3-2024-report.aspx (accessed on 19 February 2025).
- Available online: https://ir.ionispharma.com/news-releases/news-release-details/ionis-pharmaceuticals-hold-technology-webcast (accessed on 19 February 2025).
- Ranasinghe, P.; Addison, M.L.; Dear, J.W.; Webb, D.J. Small interfering RNA: Discovery, pharmacology and clinical development-An introductory review. Br. J. Pharmacol. 2023, 180, 2697–2720. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Tantray, V.G.; Kirmani, A.R.; Ahangar, A.G. A review on current status of antiviral siRNA. Rev. Med Virol. 2018, 28, e1976. [Google Scholar] [CrossRef] [PubMed]
- Kotelianski, V.; Zatsepin, T.S.; Kotelevtsev, Y.V. Lipid nanoparticles for targeted siRNA delivery—Going from bench to bedside. Int. J. Nanomed. 2016, 11, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Khvorova, A. RNAi-based drug design: Considerations and future directions. Nat. Rev. Drug Discov. 2024, 23, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja-Narashimha, K.; Huang, C.; Cao, M.; Chen, Y.P.; Borodovsky, A.; Olson, W.C.; Morton, L.G.; Retter, M.W. Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination with cemdisiran in non-human primates. PLoS ONE 2022, 17, e0269749. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sugar Modification | Examples | Effects on siRNA |
|---|---|---|
| 2′-O-Methyl (2′-OMe) | Methyl group at the 2′ position of the ribose sugar. Naturally occurring in some RNAs |
|
| 2′-Deoxy-2′-Fluoro (2′-F) | Fluorine atom at the 2′ position of the ribose sugar. |
|
| 2′-O-Methoxyethyl (2′-O-MOE) | 2-Methoxyethyl group at the 2′ position of the ribose sugar. |
|
| 2′-Arabino-Fluoro (2′-Ara-F) | Fluorine at the 2′ position with a different sugar configuration than 2′F |
|
| 2′-O-Benzyl & 2′-O-CH2Py |
| |
| Locked Nucleic Acid (LNA) | Methylene bridge connecting the 2′-O and 4′-C of the ribose, “locking” the sugar in a specific conformation |
|
| Backbone Modifications | ||
| Phosphorothioate | Replacements of the phosphate backbone |
|
| Phosphorodithioate (PS2) |
| |
| Methylphosphonate (MP) and Methoxypropylphosphonate (MOP) |
| |
| Peptide Nucleic Acid (PNA) | Replacement of the ribose phosphate backbone with a peptide backbone |
|
| Phosphonate Analog |
| |
| Base Modifications | ||
| 2-thiouridine S2U |
| |
| N-Ethylpiperidine triazole |
| |
| 6′-Phenylpyrrolocytosine (PhpC) |
| |
| 2,4-Difluorotolylribonucleoside (rF) |
| |
| 5-Nitroindole Modification |
| |
| 5-Fluoro-2′-Deoxyuridine (FdU) |
| |
| GalNAc | Lipid-Based | Naked siRNA | References | |
|---|---|---|---|---|
| Tri-GalNAc (N-acetylgalactosamine) Conjugation | Tri-GalNAc is a small sugar molecule that can be conjugated to siRNA molecules. It facilitates the targeting of siRNA, particularly to hepatocytes, which specify a high level of the asialoglycoprotein receptor (ASGPR) on their surface. Tri-GalNAc conjugation enhances siRNA delivery to liver cells, increasing efficacy and reducing off-target effects. | Lipid-based and naked siRNA delivery systems do not rely on tri-GalNAc conjugation for specific targeting of hepatocytes. Instead, they can deliver siRNA to various cell types through different mechanisms. | [81,103,104,105] | |
| Structure | Tri-GalNAc is typically conjugated to the 3′-end of the sense strand siRNA molecule or other delivery vehicles, such as lipid nanoparticles. | Lipid-based siRNA delivery systems contain lipid nanoparticles that encapsulate and protect siRNA molecules. The lipid nanoparticles can be composed of cationic lipids, which help to form stable complexes with siRNA or other lipid components designed to enhance delivery and cellular uptake. | Naked siRNA molecules that are not encapsulated or protected by any delivery system. They are typically administered as free siRNA molecules. | [8,106,107,108,109,110,111,112] |
| Target cell specificity | Tri-GalNAc conjugation allows for specific targeting of siRNA to hepatocytes in the liver by binding to the ASGPR receptor. This targeting improves siRNA’s cellular uptake and efficacy in liver-related diseases, such as hepatitis and liver cancer. | Lipid-based and naked siRNA delivery systems do not possess inherent target cell specificity, and the cellular uptake and distribution of siRNA rely on factors like nanoparticle properties, formulation, and route of administration. The lipid nanoparticles are often modified by incorporating targeting ligands (e.g., antibodies, aptamers) that specifically bind to receptors on the surface of desired target cells | [106,113,114,115,116,117,118,119,120,121,122] | |
| Stability and cellular uptake efficiency | Chemical modification significantly improves the metabolic stability of siRNA and enhances cellular uptake, specifically in hepatocytes, leading to improved siRNA efficacy in the liver when conjugated to tri-GalNAc for targeted delivery to hepatocytes | Lipid nanoparticles protect siRNA against degradation and facilitate efficient cellular uptake, increasing siRNA stability and higher delivery efficiency. | Naked siRNA is typically less stable and has lower cellular uptake efficiency due to its susceptibility to degradation and limited ability to enter cells. | [123,124,125,126,127] |
| Compounds | NCT Number | Study Title | Study Status | Conditions | Interventions | Sponsor | Phases | Study Type | Completion Date |
|---|---|---|---|---|---|---|---|---|---|
| LY3819469 | NCT05932446 | A Study to Compare Two Formulations of LY3819469 in Healthy Participants | Completed | Healthy | DRUG: LY3819469 | Eli Lilly and Company | PHASE1 | Interventional | 13 November 2023 |
| NCT05841277 | A Study of LY3819469 in Participants with Impaired and Normal Renal Function | Completed | Renal Insufficiency Healthy | DRUG: LY3819469 | Eli Lilly and Company | PHASE1 | Interventional | 4 March 2024 | |
| NCT05936138 | A Study of LY3502970 in Participants with Normal and Impaired Renal Function | Completed | Renal Insufficiency Healthy | DRUG: LY3502970 | Eli Lilly and Company | PHASE1 | Interventional | 21 June 2024 | |
| NCT04914546 | A Study of LY3819469 in Healthy Participants | Completed | Healthy | DRUG: LY3819469 DRUG: Placebo | Eli Lilly and Company | PHASE1 | Interventional | 9 November 2022 | |
| NCT05565742 | A Study of LY3819469 in Participants with Elevated Lipoprotein(a) [Lp(a)] | Completed | Lipoprotein Disorder | DRUG: LY3819469 DRUG: Placebo | Eli Lilly and Company | PHASE2 | Interventional | 18 October 2024 | |
| NCT06292013 | A Study to Investigate the Effect of Lepodisiran on the Reduction in Major Adverse Cardiovascular Events in Adults With Elevated Lipoprotein(a) - ACCLAIM-Lp(a) | Recruiting | Atherosclerotic Cardiovascular Disease (ASCVD) Elevated Lp(a) | Drug: Lepodisiran Sodium Drug: Placebo | Eli Lilly and Company | PHASE 3 | Interventional | March 2029 | |
| LY3561774 | NCT04644809 | A Study of LY3561774 in Participants with Dyslipidemia | Completed | Dyslipidemias | DRUG: LY3561774 DRUG: Placebo | Eli Lilly and Company | PHASE1 | Interventional | 17 May 2022 |
| NCT05256654 | A Study of LY3561774 in Participants with Mixed Dyslipidemia | Completed |
| DRUG: LY3561774 DRUG: Placebo | Eli Lilly and Company | PHASE2 | Interventional | 3 May 2024 | |
| DCR-AUD | NCT05021640 | Study of DCR-AUD in Healthy Volunteers | Completed | Alcohol Use Disorder (AUD) | DRUG: DCR-AUD DRUG: Placebo for DCR-AUD | Dicerna Pharmaceuticals, Inc., a Novo Nordisk company | PHASE1 | Interventional | 31 December 2022 |
| NCT05845398 | Phase 1b Study of DCR-AUD in Healthy Volunteers | Completed | Alcohol Use Disorder | DRUG: DCR-AUD DRUG: Placebo | Dicerna Pharmaceuticals, Inc., a Novo Nordisk company | PHASE1 | Interventional | 22 August 2023 | |
| ARO-HSD | NCT04202354 | Study of ARO-HSD in Healthy Volunteers and Patients with Non-Alcoholic Steatohepatitis (NASH) or Suspected NASH | Completed | Non-alcoholic Steatohepatitis | DRUG: ARO-HSD Injection DRUG: sterile normal saline (0.9% NaCl) | Arrowhead Pharmaceuticals | PHASE1 | Interventional | 23 November 2021 |
| JNJ-75220795 | NCT05039710 | A Study of JNJ-75220795 in Japanese Participants | Terminated | Fatty Liver | DRUG: JNJ-75220795 OTHER: Placebo | Janssen Pharmaceutical K.K. | PHASE1 | Interventional | 22 February 2023 |
| NCT04844450 | A Single and Multiple Ascending Dose Study of Subcutaneously Administered JNJ-75220795 | Completed | Fatty Liver Disease | DRUG: JNJ-75220795 DRUG: Placebo | Janssen Research & Development, LLC | PHASE1 | Interventional | 31 March 2023 | |
| ARO-MUC5AC | NCT05292950 | Study of ARO-MUC5AC in Healthy Subjects and Patients with Muco-Obstructive Lung Disease | Terminated |
| DRUG: ARO-MUC5AC DRUG: Placebo | Arrowhead Pharmaceuticals | PHASE1 | Interventional | 12 November 2024 |
| ARO-MMP7 | NCT05537025 | Study of ARO-MMP7 Inhalation Solution in Healthy Subjects and Patients with Idiopathic Pulmonary Fibrosis | Recruiting | Idiopathic Pulmonary Fibrosis | DRUG: ARO-MMP7 Inhalation Solution DRUG: Placebo | Arrowhead Pharmaceuticals | PHASE1 PHASE2 | Interventional | March 2025 |
| ARO-RAGE | NCT05533294 | Study of ARO-RAGE in Healthy Subjects | Completed | Asthma | DRUG: ARO-RAGE Injection DRUG: Placebo | Arrowhead Pharmaceuticals | PHASE1 | Interventional | 8 February 2024 |
| NCT05276570 | Study of ARO-RAGE in Healthy Subjects and Patients with Asthma | Active, not recruiting | Inflammatory Lung Disease | DRUG: ARO-RAGE DRUG: Placebo | Arrowhead Pharmaceuticals | PHASE1 | Interventional | March 2025 | |
| HZN-457 | NCT05565768 | Study to Evaluate HZN-457 in Healthy Volunteers | Completed | Healthy | DRUG: HZN-45 DRUG: Placebo | Horizon Therapeutics Ireland DAC | PHASE1 | Interventional | 9 August 2023 |
| ARO-C3 | NCT05083364 | Study of ARO-C3 in Adult Healthy Volunteers and Patients with Complement Mediated Renal Disease | Active, not recruiting |
| DRUG: ARO-C3DRUG: Placebo | Arrowhead Pharmaceuticals | PHASE1 | Interventional | June 2025 |
| ALN-APP | NCT05231785 | A Study to Evaluate the Safety and Tolerability of ALN-APP in Patients With EOAD | Recruiting | Early-Onset Alzheimer Disease | DRUG: ALN-APP DRUG: Placebo | Alnylam Pharmaceuticals | PHASE1 | Interventional | July 2025 |
| NCT06393712 | A Phase 2 Trial of ALN-APP in Patients with Cerebral Amyloid Angiopathy | Recruiting |
| DRUG: Placebo DRUG: ALN-APP | Alnylam Pharmaceuticals | Phase 2 | Interventional | 1 November 2029 | |
| ALN-XDH | NCT05256810 | A Study to Evaluate ALN-XDH in Healthy Subjects and Patients with Gout | Terminated | Gout | DRUG: ALN-XDH DRUG: Placebo | Alnylam Pharmaceuticals | PHASE1 PHASE2 | Interventional | 25 January 2023 |
| ALN-HSD | NCT05519475 | A Study to Evaluate the Efficacy and Safety of ALN-HSD in Adult Participants with Non-alcoholic Steatohepatitis (NASH) With Fibrosis with Genetic Risk Factors | Recruiting | Nonalcoholic Steatohepatitis | DRUG: ALN-HSD DRUG: Placebo | Regeneron Pharmaceuticals | PHASE2 | Interventional | 8 September 2027 |
| NCT04565717 | A Study of ALN-HSD in Healthy Adult Subjects and Adult Patients with Nonalcoholic Steatohepatitis (NASH) | Terminated | Non-alcoholic Steatohepatitis NASH | DRUG: ALN-HSD DRUG: Placebo | Alnylam Pharmaceuticals | PHASE1 | Interventional | 21 December 2023 | |
| ALN-KHK | NCT05761301 | A Phase 1/2 Study to Evaluate ALN-KHK in Overweight to Obese Healthy Volunteers and Obese Patients with T2DM | Active, not recruiting | Type 2 Diabetes Mellitus (T2DM) | DRUG: ALN-KHK DRUG: Placebo | Alnylam Pharmaceuticals | PHASE1PHASE2 | Interventional | 30 April 2025 |
| JNJ-73763989 | NCT05005507 | A Study of JNJ-73763989, Pegylated Interferon Alpha-2a, and Nucleos(t)Ide Analogs in Participants with Chronic Hepatitis B Virus Infection | Terminated | Hepatitis B, Chronic | DRUG: JNJ-73763989 DRUG: PegIFN-alpha-2a DRUG: Tenofovir disoproxil DRUG: TAF DRUG: ETV | Janssen Research & Development, LLC | PHASE2 | Interventional | 29 December 2021 |
| NCT04667104 | A Study of JNJ-73763989, JNJ-56136379, Nucleos(t)Ide Analogs, and Pegylated Interferon Alpha-2a in Virologically Suppressed Participants with Chronic Hepatitis B Virus Infection | Completed | Hepatitis B, Chronic | DRUG: JNJ-73763989 DRUG: Tenofovir disoproxil DRUG: Tenofovir alafenamide (TAF) DRUG: Entecavir (ETV) monohydrate DRUG: PegIFN-alpha2a | Janssen Research & Development, LLC | PHASE2 | Interventional | 17 April 2023 | |
| NCT04002752 | A Study of JNJ-73763989 in Healthy Japanese Adult Participants | Completed | Healthy | DRUG: JNJ-73763989 DRUG: Placebo | Janssen Sciences Ireland UC | PHASE1 | Interventional | 23 August 2019 | |
| NCT04586439 | A Study of JNJ-73763989 in Healthy Chinese Adult Participants | Completed | Healthy | DRUG: JNJ-73763989 | Janssen Research & Development, LLC | PHASE1 | Interventional | 18 February 2021 | |
| NCT04439539 | A Study of JNJ-73763989, Pegylated Interferon Alpha-2a, Nucleos(t)Ide Analog (NA) With or Without JNJ-56136379 in Treatment-naive Participants with Hepatitis B e Antigen (HBeAg) Positive Chronic Hepatitis B Virus (HBV) Infection | Completed | Hepatitis B, Chronic | DRUG: JNJ-73763989 DRUG: PegIFN-alpha-2a DRUG: Tenofovir disoproxil DRUG: Tenofovir alafenamide DRUG: JNJ-56136379 | Janssen Research & Development, LLC | PHASE2 | Interventional | 13 February 2024 | |
| NCT04129554 | A Study of JNJ 73763989 + JNJ 56136379 + Nucleos(t)Ide Analog (NA) Regimen Compared to NA Alone in e Antigen Negative Virologically Suppressed Participants with Chronic Hepatitis B Virus Infection | Completed | Hepatitis B, Chronic | DRUG: JNJ-73763989 DRUG: JNJ-56136379 DRUG: Placebo for JNJ-73763989 DRUG: Placebo for JNJ-56136379 DRUG: Entecavir (ETV) monohydrate DRUG: Tenofovir disoproxil fumarate (TDF) DRUG: Tenofovir alafenamide (TAF) | Janssen Sciences Ireland UC | PHASE2 | Interventional | 9 June 2022 | |
| NCT05275023 | An Efficacy and Safety Study of a Combination of JNJ-73763989, Nucleos(t)Ide Analogs (NAs), and a Programmed Cell Death Protein Receptor-1 (PD-1) Inhibitor in Chronic Hepatitis B Participants | Completed | Hepatitis B, Chronic | DRUG: JNJ-73763989 DRUG: PD-1 inhibitor DRUG: Tenofovir Disoproxil DRUG: Tenofovir Alafenamide DRUG: Entecavir | Janssen Research & Development, LLC | PHASE2 | Interventional | 31 May 2024 | |
| NCT04535544 | A Study of JNJ-73763989 + Nucleos(t)Ide Analog in Participants Co-Infected With Hepatitis B and Hepatitis D Virus | Active not recruiting | Hepatitis D, Chronic | DRUG: JNJ-73763989 DRUG: Placebo DRUG: Entecavir (ETV) monohydrate DRUG: Tenofovir disoproxil DRUG: Tenofovir alafenamide (TAF) | Janssen Research & Development, LLC | PHASE2 | INTERVENTIONAL | 6 March 2025 | |
| NCT04963738 | A Study of JNJ-73763989 in Adult Participants With Renal Impairment | Completed | Renal Impairment | DRUG: JNJ-73763989 | Janssen Research & Development, LLC | PHASE1 | Interventional | 17 October 2022 | |
| NCT04208386 | A Study to Evaluate the Effect of Hepatic Impairment on JNJ-73763989 | Completed | Hepatic Impairment | DRUG: JNJ-73763989 | Janssen Sciences Ireland UC | PHASE1 | Interventional | 20 July 2020 | |
| NCT03982186 | A Study of Different Combination Regimens Including JNJ-73763989 and/or JNJ-56136379 for the Treatment of Chronic Hepatitis B Virus Infection | Completed | Hepatitis B, Chronic | DRUG: JNJ-73763989 DRUG: Placebo for JNJ-73763989 DRUG: JNJ-56136379 DRUG: Placebo for JNJ-56136379 DRUG: Nucleos(t)ide Analog (NA) | Janssen Sciences Ireland UC | PHASE2 | Interventional | 26 April 2022 | |
| NCT04585789 | A Study to Assess Intrahepatic and Peripheral Changes in Immunologic and Virologic Markers in Chronic Hepatitis B Virus Infection | Completed | Hepatitis B | DRUG: JNJ-73763989 DRUG: JNJ-56136379 DRUG: Entecavir (ETV)|DRUG: Tenofovir disoproxil DRUG: Tenofovir alafenamide (TAF) DRUG: PegIFN-alpha-2a (Optional) | Janssen Research & Development, LLC | PHASE2 | Interventional | 9 January 2024 | |
| NCT05123599 | A Study of JNJ-73763989, JNJ-64300535, and Nucleos(t)Ide Analogs in Virologically Suppressed, Hepatitis B e Antigen (HBeAg)- Negative Participants with Chronic Hepatitis B Virus Infection | Completed | Hepatitis B, Chronic | DRUG: JNJ-73763989 BIOLOGICAL: JNJ-64300535 DRUG: ETV monohydrate DRUG: Tenofovir disoproxil DRUG: TAF | Janssen Research & Development, LLC | PHASE1 | Interventional | 26 June 2024 | |
| VIR-2218 | NCT04749368 | Study to Investigate the Safety and Efficacy of BRII-835 and BRII-179 Combination Therapy Treating Chronic HBV Infection | Completed | Hepatitis B, Chronic | DRUG: BRII-835 (VIR-2218) BIOLOGICAL: BRII-179 (VBI-2601) with IFN-α BIOLOGICAL: BRII-179 (VBI-2601) | Brii Biosciences Limited | PHASE2 | Interventional | 24 July 2023 |
| NCT05484206 | Effect of Hepatic Impairment on the PK and Safety of VIR-2218 and VIR-3434 | Recruiting | Hepatic Impairment|Cirrhosis | DRUG: VIR-2218 DRUG: VIR-3434 | Vir Biotechnology, Inc. | PHASE1 | Interventional | 25 September 2026 | |
| NCT04412863 | Study of VIR-2218 With or Without Pegylated Interferon Alpha-2a for Treatment of Chronic Hepatitis B Virus Infection | Completed | Chronic Hepatitis B | DRUG: VIR-2218 DRUG: pegylated interferon-alfa 2a | Vir Biotechnology, Inc. | PHASE2 | Interventional | 25 March 2024 | |
| NCT04507269 | Study of VIR-2218 in Patients With Chronic Hepatitis B in Mainland China | Completed | Hepatitis B, Chronic | DRUG: VIR-2218 DRUG: Placebo | Brii Biosciences Limited | PHASE2 | Interventional | 30 September 2021 | |
| NCT05612581 | A Platform Study to Evaluate Investigational Therapies in Chronic Hepatitis B Infection | Active, not recruiting | Hepatitis B, Chronic | DRUG: VIR-3434 DRUG: VIR-2218 DRUG: TDF DRUG: PEG-IFNα | Vir Biotechnology, Inc. | PHASE1|PHASE2 | Interventional | March 2027 | |
| NCT05970289 | Investigate the Efficacy and Safety of BRII-835 (VIR-2218) and PEG-IFNα Combination Therapy in Chronic HBV Patients | Active, not recruiting | Chronic Hepatitis B Virus Infection | BIOLOGICAL: PEG-IFNα DRUG: BRII-835 | Brii Biosciences Limited | PHASE2 | Interventional | February 2026 | |
| NCT03672188 | Study of VIR-2218 in Healthy Subjects and Patients With Chronic Hepatitis B | Completed | Chronic Hepatitis B | DRUG: VIR-2218 DRUG: Placebo | Vir Biotechnology, Inc. | PHASE1|PHASE2 | Interventional | 3 September 2020 | |
| NCT04856085 | Study of VIR-2218, VIR-3434, and/or PEG-IFNα in Subjects With Chronic Hepatitis B Virus Infection | Active, not recruiting | Hepatitis B, Chronic | DRUG: VIR-2218 DRUG: VIR-3434 DRUG: PEG-IFNα | Vir Biotechnology, Inc. | PHASE2 | Interventional | June 2027 | |
| NCT05844228 | A Study to Investigate the Effect of Renal Impairment on the PK and Safety of VIR-2218 | Recruiting | Renal Impairment | DRUG: VIR-2218 | Vir Biotechnology, Inc. | PHASE1 | Interventional | 21 November 2025 | |
| NCT05461170 | SOLSTICE: Combination Therapy for the Treatment of Chronic Hepatitis D Infection | Recruiting | Hepatitis D, Chronic | DRUG: VIR-2218 DRUG: VIR-3434 DRUG: NRTI | Vir Biotechnology, Inc. | PHASE2 | Interventional | August 2029 | |
| NCT04891770 | Study to Evaluate the Safety and Efficacy of Selgantolimod (SLGN)-Containing Combination Therapies for the Treatment of Chronic Hepatitis B (CHB) | Completed | Chronic Hepatitis B | DRUG: Tenofovir Alafenamide DRUG: VIR-2218 DRUG: Nivolumab DRUG: Selgantolimod | Gilead Sciences | PHASE2 | Interventional | 19 July 2024 | |
| AB-729 | NCT04980482 | Open-Label Study of AB-729, Nucleos(t)Ide Analogue and Pegylated Interferon Alfa-2a in Subjects with Chronic Hepatitis B Infection | Active, not recruiting | Chronic Hepatitis b | DRUG: AB-729 DRUG: Peg-IFNα-2a | Arbutus Biopharma Corporation | PHASE 2 | Interventional | April 2025 |
| NCT04820686 | A Study Evaluating Treatment Regimens Containing Vebicorvir (ABI-H0731) in Participants with Chronic Hepatitis B Infection | Terminated | Chronic Hepatitis B | DRUG: VBR DRUG: AB-729 DRUG: SOC NrtI | Assembly Biosciences Arbutus Biopharma Corporation | PHASE 2 | Interventional | 30 March 2023 | |
| NCT04847440 | A Study of Safety and Efficacy of ATI-2173 in Combination with Tenofovir Disoproxil Fumarate in Subjects with Chronic Hepatitis B Virus Infection and Subjects with Hepatitis D Virus Coinfection | Terminated | Hepatitis B, Chronic Hepatitis D | DRUG: ATI-2173 DRUG: Viread DRUG: AB-729 | Antios Therapeutics, Inc. | PHASE 2 | Interventional | 1 September 2022 | |
| NCT06277037 | Long-Term Follow-up Study for Subjects With CHB Previously Treated With Imdusiran (AB729) | Recruiting | Long Term Follow-up | Other: Non-interventional | Arbutus Biopharma Corporation | Phase 2 | Observational | 30 October 2029 | |
| NCT06154278 | Intrahepatic and Peripheral Responses to Imdusiran (AB-729) in Chronic Hepatitis B | Recruiting | Chronic Hepatitis B | DRUG: Imdusiran (AB-729) | University of Maryland, Baltimore | Phase 2 | Interventional | 1 July 2027 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebenezer, O.; Oyebamiji, A.K.; Olanlokun, J.O.; Tuszynski, J.A.; Wong, G.K.-S. Recent Update on siRNA Therapeutics. Int. J. Mol. Sci. 2025, 26, 3456. https://doi.org/10.3390/ijms26083456
Ebenezer O, Oyebamiji AK, Olanlokun JO, Tuszynski JA, Wong GK-S. Recent Update on siRNA Therapeutics. International Journal of Molecular Sciences. 2025; 26(8):3456. https://doi.org/10.3390/ijms26083456
Chicago/Turabian StyleEbenezer, Oluwakemi, Abel Kolawole Oyebamiji, John Oludele Olanlokun, Jack A. Tuszynski, and Gane Ka-Shu Wong. 2025. "Recent Update on siRNA Therapeutics" International Journal of Molecular Sciences 26, no. 8: 3456. https://doi.org/10.3390/ijms26083456
APA StyleEbenezer, O., Oyebamiji, A. K., Olanlokun, J. O., Tuszynski, J. A., & Wong, G. K.-S. (2025). Recent Update on siRNA Therapeutics. International Journal of Molecular Sciences, 26(8), 3456. https://doi.org/10.3390/ijms26083456