
Exploring Morphological, Transcriptomic, and Metabolomic Differences Between Two Sister Lines with Contrasting Resistance to Orange Rust Disease in Sugarcane

 , ,
, , 

Abstract
:1. Introduction
2. Results
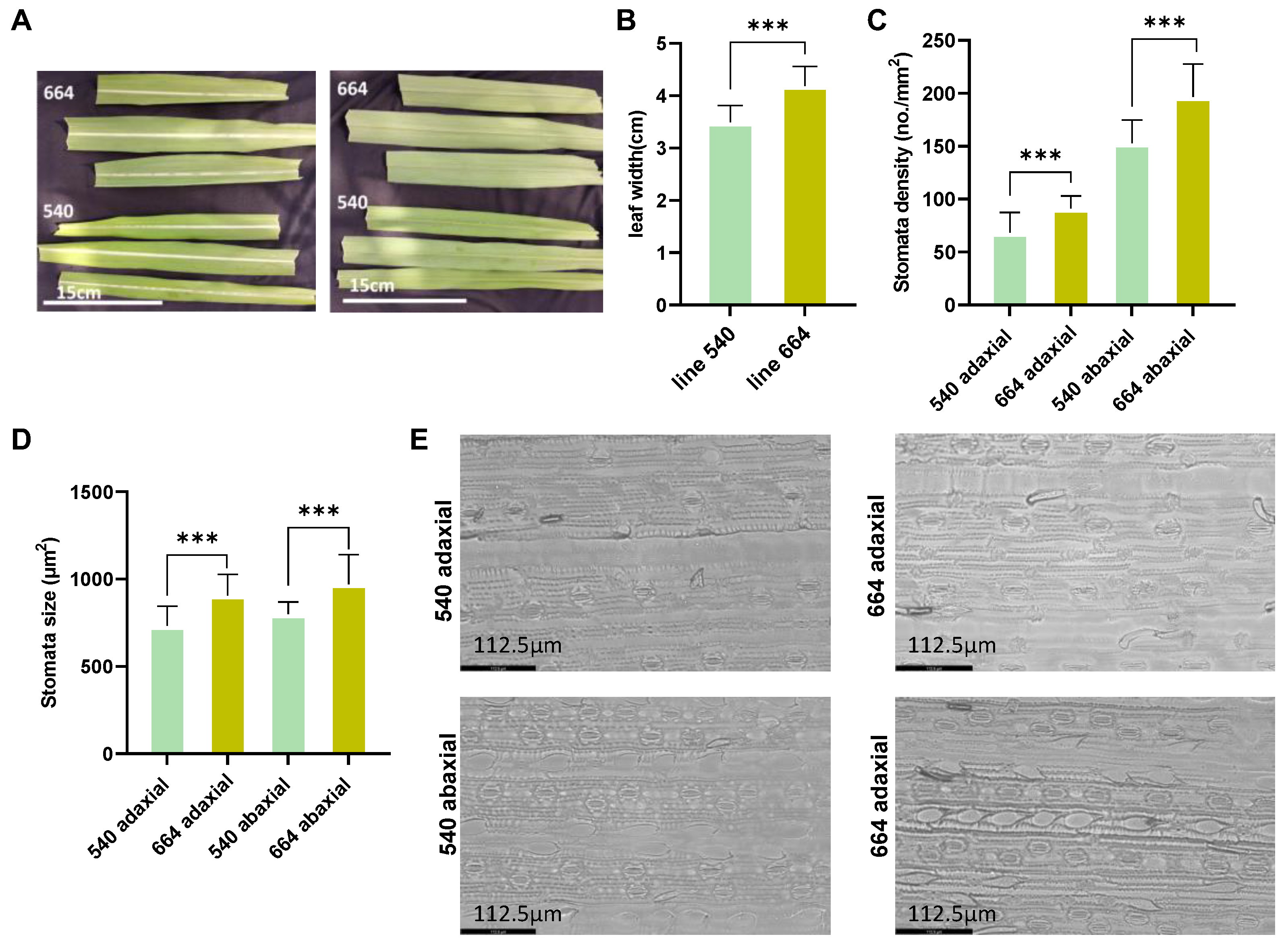
2.1. Morphological Observations
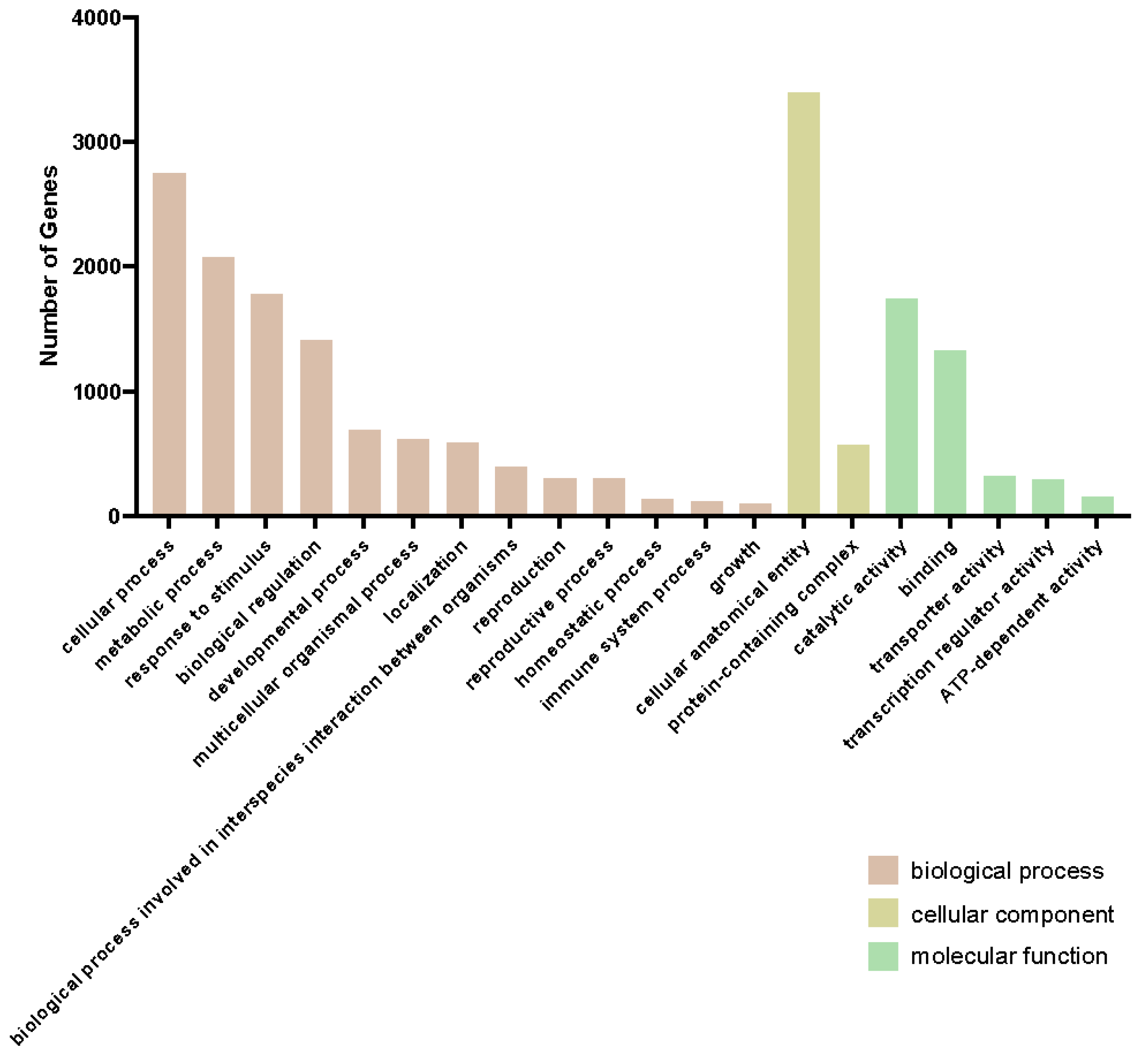
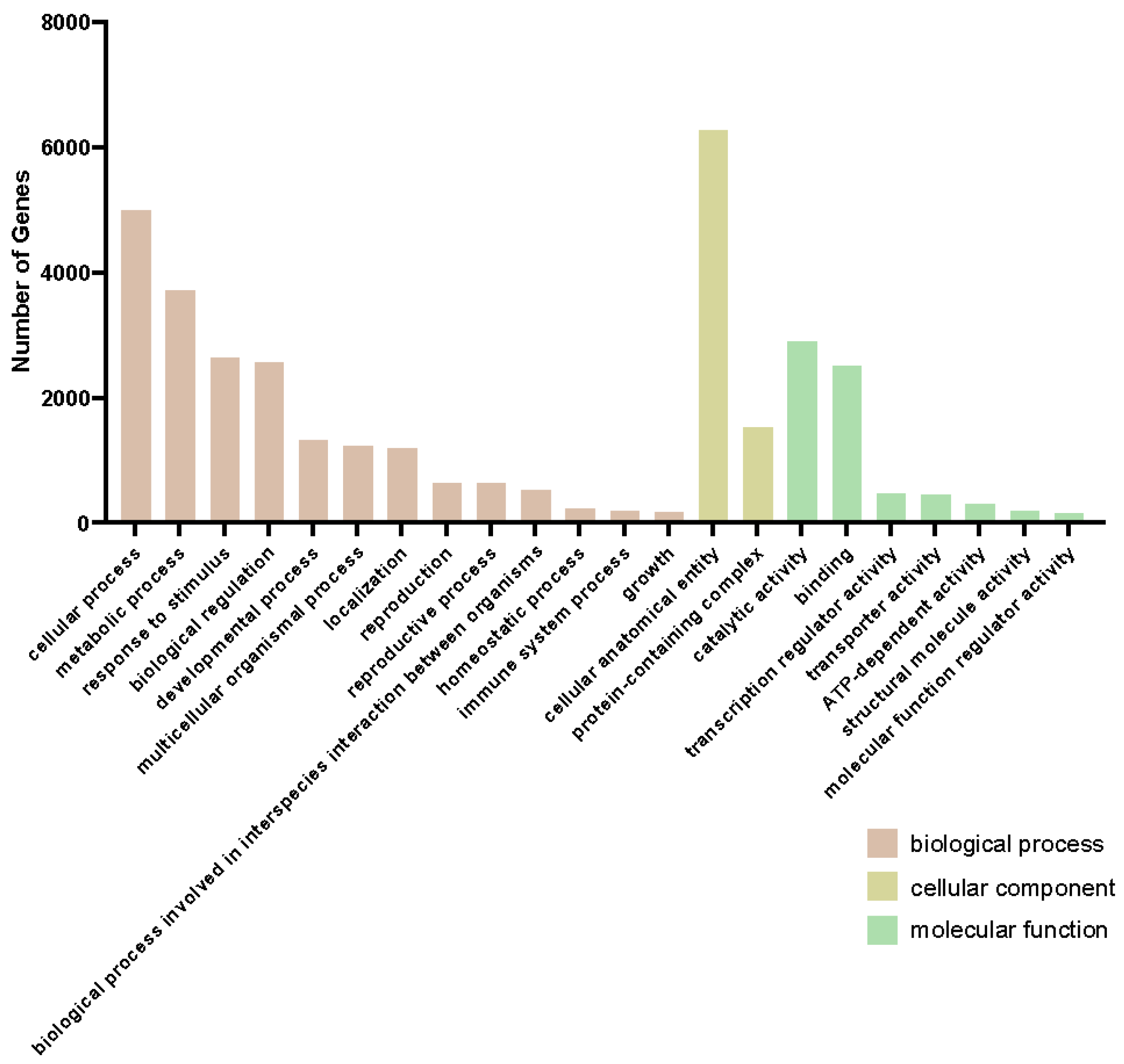
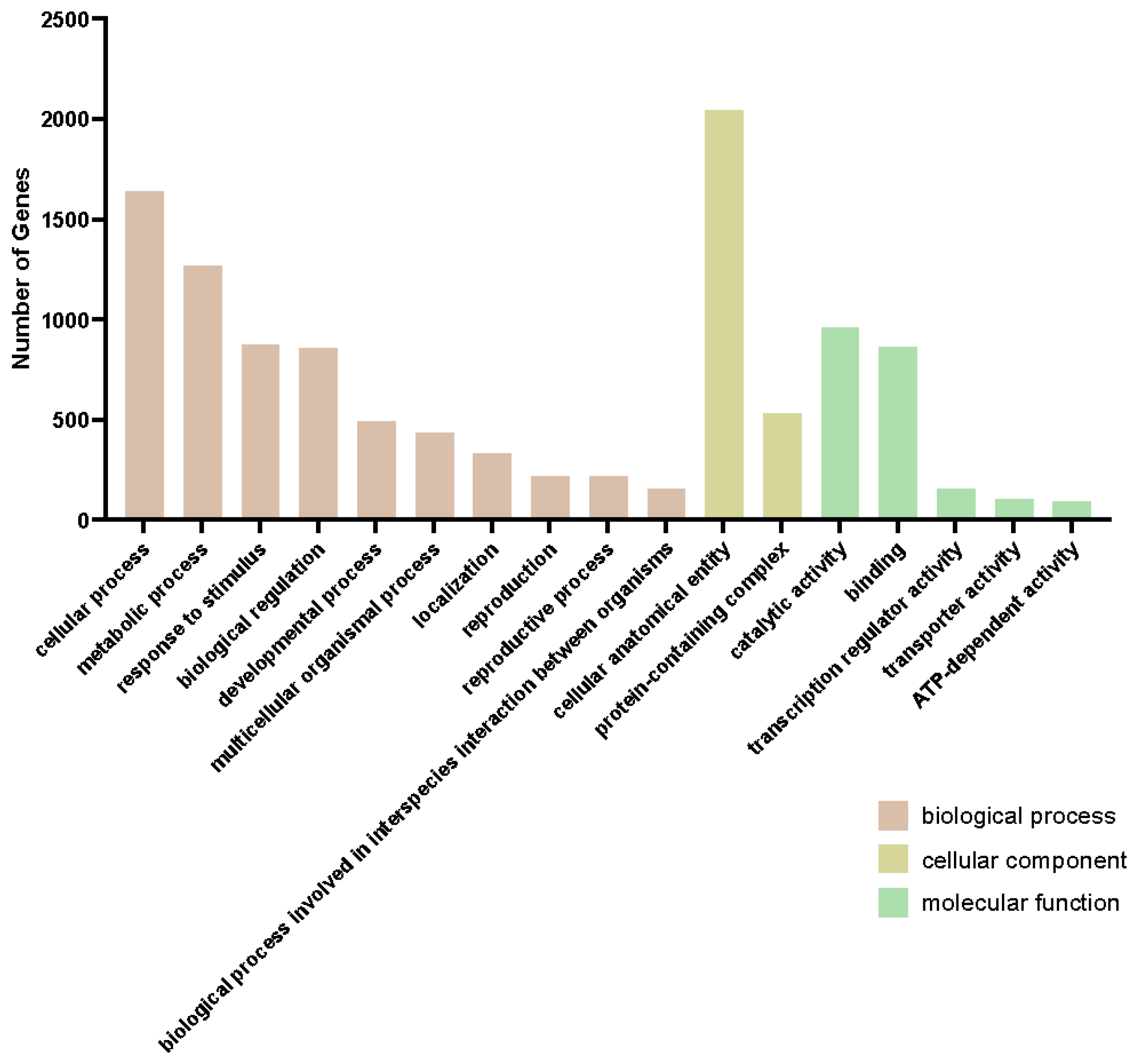
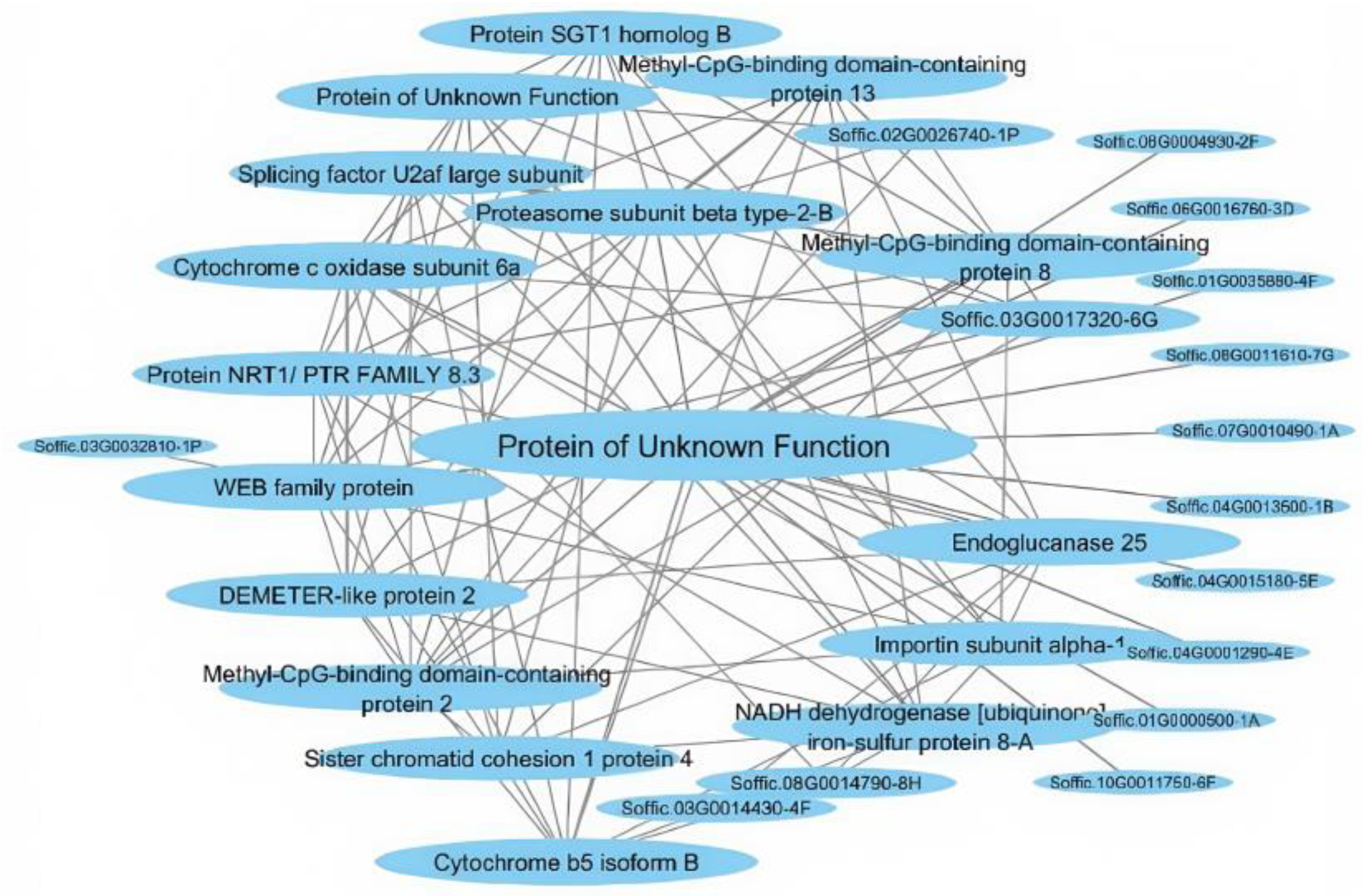
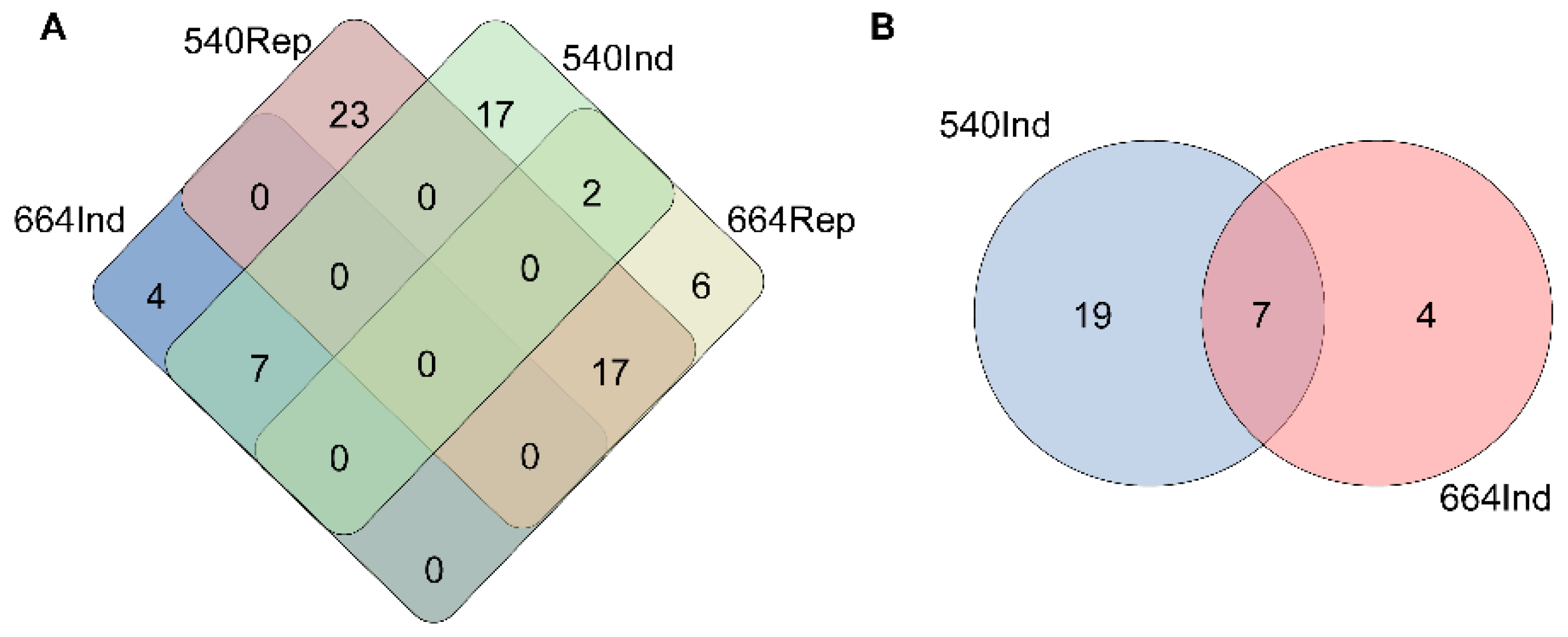
2.2. mRNAseq Analysis
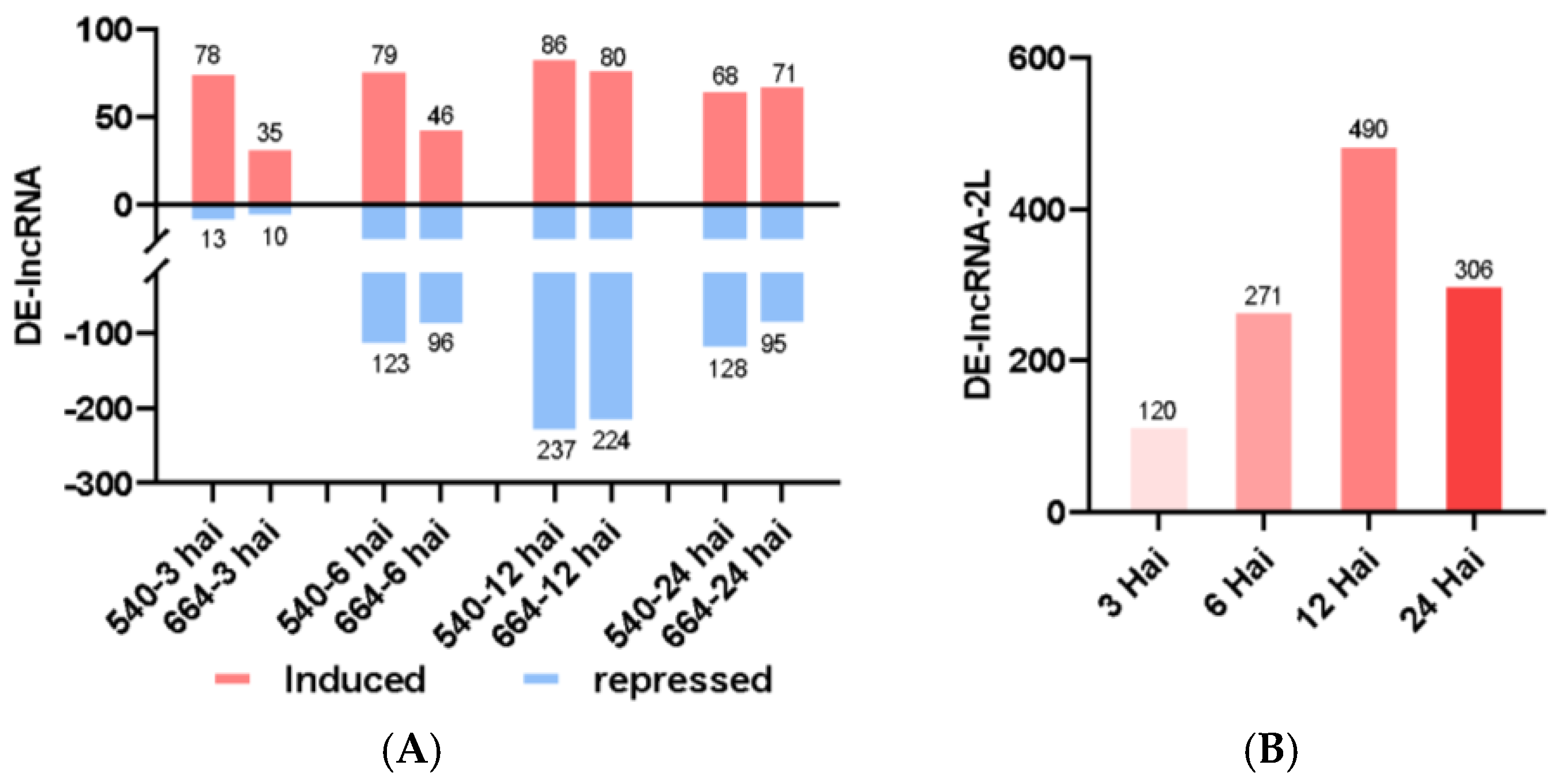
2.3. Long Non-Coding RNA (lncRNA) Analysis
2.4. sRNAseq Analysis
2.5. Weighted Correlation Network Analysis (WGCNA)
2.6. Detection of Genomic Variation
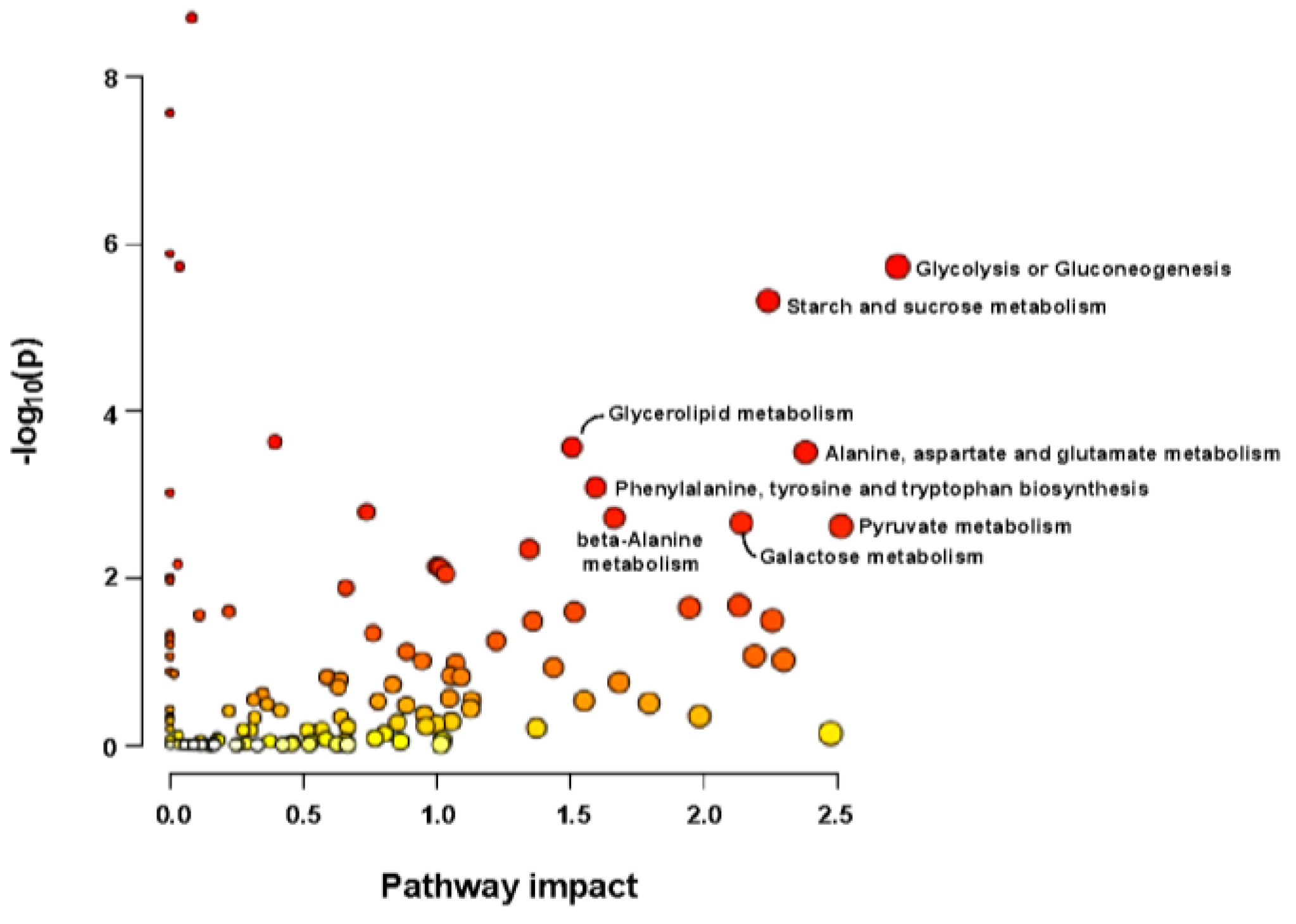
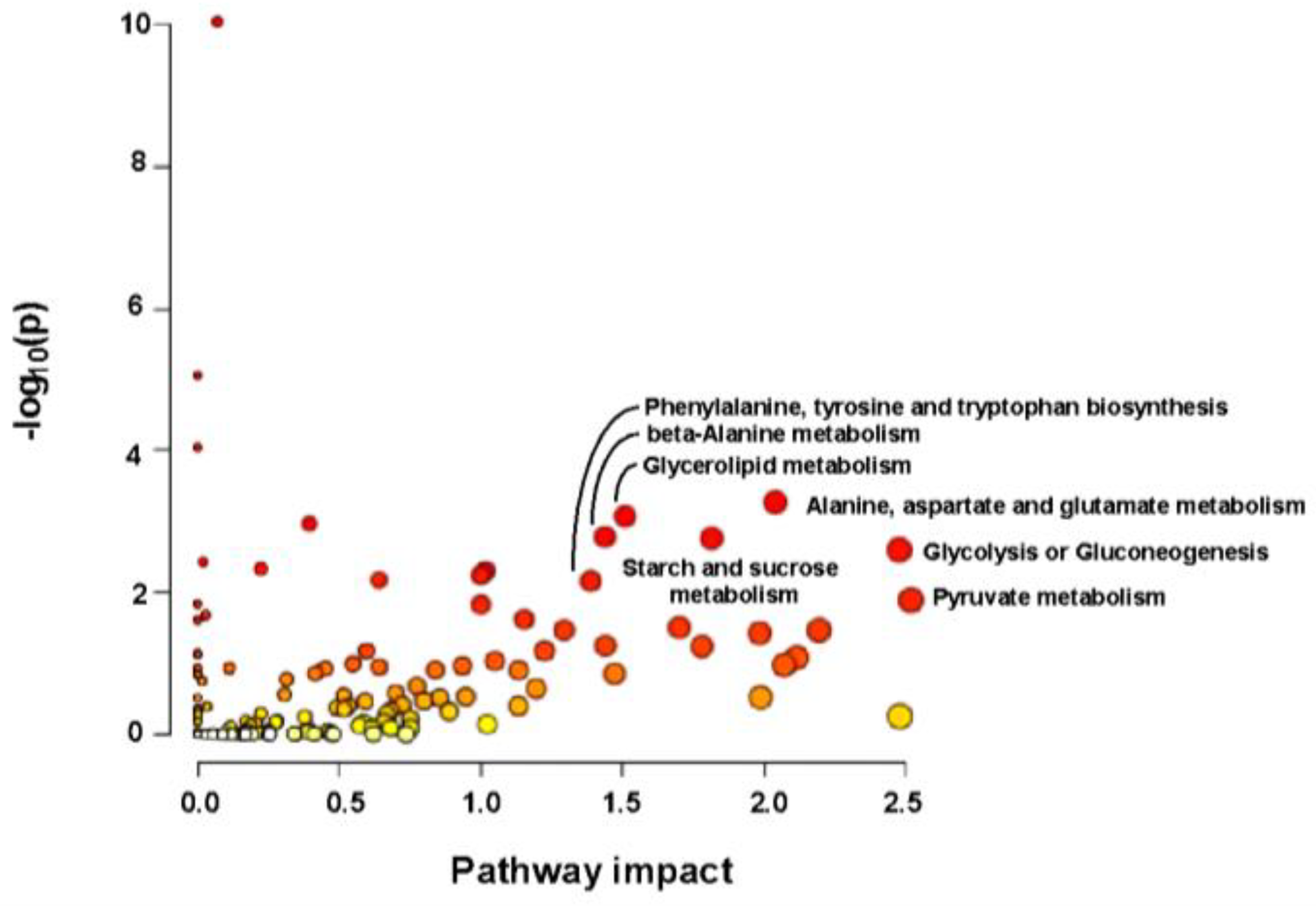
2.7. Metabolomic Analysis
2.8. Transcriptional and Metabolomic Joint Analysis
3. Discussion
3.1. Morphological Analysis
3.2. Transcriptomic Analysis
3.3. Metabolomic Analysis
3.4. Limitations and Future Directions
4. Materials and Methods
4.1. Plant Materials
4.2. Sugarcane Inoculation
4.3. Morphological Observation
4.4. RNAseq Library Construction and Sequencing
4.5. mRNAseq Analysis
- If a gene was a DEG in one line at a time point but not expressed or not a DEG in the other line, then this gene was defined as a DEG2L. (This DEG2L in this scenario suggests that a particular DEG undergoes a significant change in expression in only one of the lines.)
- If a gene was expressed in both lines and was a DEG of either line at a time point, but the FC values (the fold change of gene expression between the given time point and 0 hai) between the two lines were different by more than three times, or the DEG had different expression trends between two lines, then this gene was defined as a DEG2L. (The DEG2L in this scenario suggests that a certain DEG exhibits a greater magnitude of change in one line, or exhibits opposing trends of change between the two lines.)
4.6. LncRNA Data Analysis
4.7. sRNAseq Data Analysis
4.8. Weighted Correlation Network Analysis (WGCNA)
4.9. SNP Identification
4.10. Metabolomic Analysis
4.11. Transcriptional and Metabolomic Joint Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piperidis, G.; Piperidis, N.; D’Hont, A. Molecular Cytogenetic Investigation of Chromosome Composition and Transmission in Sugarcane. Mol. Genet. Genom. 2010, 284, 65–73. [Google Scholar] [CrossRef]
- Yadav, S.; Jackson, P.; Wei, X.; Ross, E.M.; Aitken, K.; Deomano, E.; Atkin, F.; Hayes, B.J.; Voss-Fels, K.P. Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection. Agronomy 2020, 10, 585. [Google Scholar] [CrossRef]
- Bordonal, R.d.O.; Carvalho, J.L.N.; Lal, R.; de Figueiredo, E.B.; de Oliveira, B.G.; La Scala, N. Sustainability of Sugarcane Production in Brazil. A Review. Agron. Sustain. Dev. 2018, 38, 13. [Google Scholar] [CrossRef]
- Liu, H.; Yang, X.; You, Q.; Song, J.; Wang, L.; Zhang, J.; Deng, Z.; Ming, R.; Wang, J. Pedigree, Marker Recruitment, and Genetic Diversity of Modern Sugarcane Cultivars in China and the United States. Euphytica 2018, 214, 48. [Google Scholar] [CrossRef]
- Sevimoğlu, O.; Rogge, W.F. Seasonal Variations of PM10—Trace Elements, PAHs and Levoglucosan: Rural Sugarcane Growing Area versus Coastal Urban Area in Southeastern Florida, USA. Part II: Elemental Concentrations. Particuology 2019, 46, 99–108. [Google Scholar] [CrossRef]
- Sugar and Sweeteners—Background. Available online: https://www.ers.usda.gov/topics/crops/sugar-and-sweeteners/background (accessed on 1 October 2024).
- Correr, F.H.; Hosaka, G.K.; Gómez, S.G.P.; Cia, M.C.; Vitorello, C.B.M.; Camargo, L.E.A.; Massola, N.S.; Carneiro, M.S.; Margarido, G.R.A. Time-Series Expression Profiling of Sugarcane Leaves Infected with Puccinia Kuehnii Reveals an Ineffective Defense System Leading to Susceptibility. Plant Cell Rep. 2020, 39, 873–889. [Google Scholar] [CrossRef]
- Xiong, H.; Chen, Y.; Gao, S.J.; Pan, Y.B.; Shi, A. Population Structure and Genetic Diversity Analysis in Sugarcane (Saccharum spp. Hybrids) and Six Related Saccharum Species. Agronomy 2022, 12, 412. [Google Scholar] [CrossRef]
- Manechini, J.R.V.; da Costa, J.B.; Pereira, B.T.; Carlini-Garcia, L.A.; Xavier, M.A.; Landell, M.G.d.A.; Pinto, L.R. Unraveling the Genetic Structure of Brazilian Commercial Sugarcane Cultivars through Microsatellite Markers. PLoS ONE 2018, 13, e0195623. [Google Scholar] [CrossRef]
- Thirugnanasambandam, P.P.; Hoang, N.V.; Henry, R.J. The Challenge of Analyzing the Sugarcane Genome. Front. Plant Sci. 2018, 9, 616. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. De Novo Assembly and Characterizing of the Culm-Derived Meta-Transcriptome from the Polyploid Sugarcane Genome Based on Coding Transcripts. Heliyon 2018, 4, e00583. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A Survey of the Complex Transcriptome from the Highly Polyploid Sugarcane Genome Using Full-Length Isoform Sequencing and de Novo Assembly from Short Read Sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Souza, G.M.; Berges, H.; Bocs, S.; Casu, R.; D’Hont, A.; Ferreira, J.E.; Henry, R.; Ming, R.; Potier, B.; Van Sluys, M.A.; et al. The Sugarcane Genome Challenge: Strategies for Sequencing a Highly Complex Genome. Trop. Plant Biol. 2011, 4, 145–156. [Google Scholar] [CrossRef]
- Vettore, A.L.; da Silva, F.R.; Kemper, E.L.; Souza, G.M.; da Silva, A.M.; Ferro, M.I.T.; Henrique-Silva, F.; Giglioti, É.A.; Lemos, M.V.F.; Coutinho, L.L.; et al. Analysis and Functional Annotation of an Expressed Sequence Tag Collection for Tropical Crop Sugarcane. Genome Res. 2003, 13, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Garsmeur, O.; Droc, G.; Antonise, R.; Grimwood, J.; Potier, B.; Aitken, K.; Jenkins, J.; Martin, G.; Charron, C.; Hervouet, C.; et al. A Mosaic Monoploid Reference Sequence for the Highly Complex Genome of Sugarcane. Nat. Commun. 2018, 9, 2638. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-Defined Genome of the Autopolyploid Sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Healey, A.L.; Garsmeur, O.; Lovell, J.T.; Shengquiang, S.; Sreedasyam, A.; Jenkins, J.; Plott, C.B.; Piperidis, N.; Pompidor, N.; Llaca, V.; et al. The Complex Polyploid Genome Architecture of Sugarcane. Nature 2024, 628, 804–810. [Google Scholar] [CrossRef]
- Bao, Y.; Zhang, Q.; Huang, J.; Zhang, S.; Yao, W.; Yu, Z.; Deng, Z.; Yu, J.; Kong, W.; Yu, X.; et al. A Chromosomal-Scale Genome Assembly of Modern Cultivated Hybrid Sugarcane Provides Insights into Origination and Evolution. Nat. Commun. 2024, 15, 3041. [Google Scholar] [CrossRef]
- Magarey, R.; Willcox, T.; Croft, B.; Cordingley, A. Orange Rust, A Major Pathogen Affecting Crops of Q124 in Queenland in 2000. In Proceedings of the 2001 Conference of the Australian Society of Sugar Cane Technologists, Mackay, Australia, 1–4 May 2001; Volume 23. [Google Scholar]
- Magarey, R.C.; Croft, B.J.; Willcox, T.G. An Epidemic of Orange Rust on Sugarcane in Australia. In Proceedings of the XXIV International Society of Sugar Cane Technologists Congress, Brisbane, Australia, 17–21 September 2001; Volume 24. [Google Scholar]
- Zhao, D.; Glynn, N.C.; Glaz, B.; Comstock, J.C.; Sood, S. Orange Rust Effects on Leaf Photosynthesis and Related Characters of Sugarcane. Plant Dis. 2011, 95, 640–647. [Google Scholar] [CrossRef]
- Comstock, J.C.; Sood, S.G.; Glynn, N.C.; Shine, J.M.; McKemy, J.M.; Castlebury, L.A. First Report of Puccinia Kuehnii, Causal Agent of Orange Rust of Sugarcane, in the United States and Western Hemisphere. Plant Dis. 2008, 92, 175. [Google Scholar] [CrossRef]
- Rott, P.; Sood, S.; Comstock, J.C.; Raid, R.N.; Glynn, N.C.; Gilbert, R.A.; Sandhu, H.S. Sugarcane Orange Rust; UF/IFAS Extension; EDIS: Gainesville, FL, USA, 2014; pp. 1–3. [Google Scholar]
- Yang, X.; Islam, M.S.; Sood, S.; Maya, S.; Hanson, E.A.; Comstock, J.; Wang, J. Identifying Quantitative Trait Loci (QTLs) and Developing Diagnostic Markers Linked to Orange Rust Resistance in Sugarcane (Saccharum spp.). Front. Plant Sci. 2018, 9, 350. [Google Scholar] [CrossRef]
- Osbourn, A.E. Preformed Antimicrobial Compounds and Plant Defense against Fungal Attack. Plant Cell 1996, 8, 1821–1831. [Google Scholar] [CrossRef]
- Melotto, M.; Underwood, W.; Koczan, J.; Nomura, K.; He, S.Y. Plant Stomata Function in Innate Immunity against Bacterial Invasion. Cell 2006, 126, 969–980. [Google Scholar] [CrossRef]
- Shafiei, R.; Hang, C.; Kang, J.G.; Loake, G.J. Identification of Loci Controlling Non-Host Disease Resistance in Arabidopsis against the Leaf Rust Pathogen Puccinia Triticina. Mol. Plant Pathol. 2007, 8, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Fang, Y.; Chen, L.; Wang, J.; Chen, X. Role of Non-Coding RNAs in Plant Immunity. Plant Commun. 2021, 2, 100180. [Google Scholar] [CrossRef] [PubMed]
- Casassola, A.; Ereful, N.C.; Zanella, C.M.; Pandey, P.; Brammer, S.P.; Chaves, M.S.; Martinelli, J.A.; Boyd, L.A. Transcriptional Profiling Identifies the Early Responses to Puccinia Triticina Infection in the Adult Plant Leaf Rust Resistant Wheat Variety Toropi. Plant Pathol. 2024, 73, 832–845. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, D.; Yin, H.; Wang, H.; Cao, C.; Wang, J.; Zheng, J.; Liu, J. Transcriptomic and Metabolomic Analyses of the Response of Resistant Peanut Seeds to Aspergillus Flavus Infection. Toxins 2023, 15, 414. [Google Scholar] [CrossRef]
- Boba, A.; Kostyn, K.; Kozak, B.; Zalewski, I.; Szopa, J.; Kulma, A. Transcriptomic Profiling of Susceptible and Resistant Flax Seedlings after Fusarium Oxysporum Lini Infection. PLoS ONE 2021, 16, e0246052. [Google Scholar] [CrossRef]
- Liu, R.; Lu, J.; Zhang, L.; Wu, Y. Transcriptomic Insights into the Molecular Mechanism of Wheat Response to Stripe Rust Fungus. Heliyon 2022, 8, e10951. [Google Scholar] [CrossRef]
- Zhang, Q.; Qi, Y.; Pan, H.; Tang, H.; Wang, G.; Hua, X.; Wang, Y.; Lin, L.; Li, Z.; Li, Y.; et al. Genomic Insights into the Recent Chromosome Reduction of Autopolyploid Sugarcane Saccharum Spontaneum. Nat. Genet. 2022, 54, 885–896. [Google Scholar] [CrossRef]
- Patel, M.K.; Maurer, D.; Feygenberg, O.; Ovadia, A.; Elad, Y.; Oren-Shamir, M.; Alkan, N. Phenylalanine: A Promising Inducer of Fruit Resistance to Postharvest Pathogens. Foods 2020, 9, 646. [Google Scholar] [CrossRef]
- Plant, M.; Hou, S.; Rodrigues, O.; Liu, Z.; Shan, L.; He, P. Small Holes, Big Impact: Stomata in Plant–Pathogen–Climate Epic Trifecta. Mol. Plant 2023, 17, 26–49. [Google Scholar] [CrossRef]
- Ahn, I.P.; Kim, S.; Lee, Y.H.; Suh, S.C. Vitamin B1-Induced Priming Is Dependent on Hydrogen Peroxide and the NPR1 Gene in Arabidopsis. Plant Physiol. 2007, 143, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Sue, M.; Horikoshi, R.; Tebayashi, S.-I.; Ishihara, A.; Endo, T.R.; Iwamura, H. Occurrence of Hordatines, the Barley Antifungal Compounds, in a Wheat-Barley Chromosome Addition Line. Genes Genet. Syst. 1999, 74, 99–103. [Google Scholar]
- Fakih, Z.; Plourde, M.B.; Germain, H. Differential Participation of Plant Ribosomal Proteins from the Small Ribosomal Subunit in Protein Translation under Stress. Biomolecules 2023, 13, 1160. [Google Scholar] [CrossRef]
- Li, G.; Chen, T.; Zhang, Z.; Li, B.; Tian, S. Roles of Aquaporins in Plant-Pathogen Interaction. Plants 2020, 9, 1134. [Google Scholar] [CrossRef] [PubMed]
- Boursiac, Y.; Lee, S.M.; Romanowsky, S.; Blank, R.; Sladek, C.; Chung, W.S.; Harper, J.F. Disruption of the Vacuolar Calcium-ATPases in Arabidopsis Results in the Activation of a Salicylic Acid-Dependent Programmed Cell Death Pathway. Plant Physiol. 2010, 154, 1158–1171. [Google Scholar] [CrossRef]
- Wimalasekera, R.; Villar, C.; Begum, T.; Scherer, G.F.E. COPPER AMINE OXIDASE1 (CuAO1) of Arabidopsis Thaliana Contributes to Abscisic Acid-and Polyamine-Induced Nitric Oxide Biosynthesis and Abscisic Acid Signal Transduction. Mol. Plant 2011, 4, 663–678. [Google Scholar] [CrossRef]
- Khan, M.; Ali, S.; Al Azzawi, T.N.I.; Yun, B.W. Nitric Oxide Acts as a Key Signaling Molecule in Plant Development under Stressful Conditions. Int. J. Mol. Sci. 2023, 24, 4782. [Google Scholar] [CrossRef]
- Zagorchev, L.; Seal, C.E.; Kranner, I.; Odjakova, M. A Central Role for Thiols in Plant Tolerance to Abiotic Stress. Int. J. Mol. Sci. 2013, 14, 7405–7432. [Google Scholar] [CrossRef]
- Boncan, D.A.T.; Tsang, S.S.K.; Li, C.; Lee, I.H.T.; Lam, H.M.; Chan, T.F.; Hui, J.H.L. Terpenes and Terpenoids in Plants: Interactions with Environment and Insects. Int. J. Mol. Sci. 2020, 21, 7382. [Google Scholar] [CrossRef]
- Razo-Belmán, R.; Ángeles-López, Y.I.; García-Ortega, L.F.; León-Ramírez, C.G.; Ortiz-Castellanos, L.; Yu, H.; Martínez-Soto, D. Fungal Volatile Organic Compounds: Mechanisms Involved in Their Sensing and Dynamic Communication with Plants. Front. Plant Sci. 2023, 14, 1257098. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.; Lii, Y.E.; Chellappan, P.; Lei, L.; Peralta, K.; Jiang, C.; Guo, J.; Coaker, G.; Jin, H. MiRNA863-3p Sequentially Targets Negative Immune Regulator ARLPKs and Positive Regulator SERRATE upon Bacterial Infection. Nat. Commun. 2016, 7, 11324. [Google Scholar] [CrossRef] [PubMed]
- Soto-Suárez, M.; Baldrich, P.; Weigel, D.; Rubio-Somoza, I.; San Segundo, B. The Arabidopsis MiR396 Mediates Pathogen-Associated Molecular Pattern-Triggered Immune Responses against Fungal Pathogens. Sci. Rep. 2017, 7, 44898. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Qi, N.; Lei, P.; Wang, Y.; Xuan, Y.; Liu, X.; Fan, H.; Chen, L.; Duan, Y.; Zhu, X. Gma-MiR408 Enhances Soybean Cyst Nematode Susceptibility by Suppressing Reactive Oxygen Species Accumulation. Int. J. Mol. Sci. 2022, 23, 14022. [Google Scholar] [CrossRef]
- Wu, J.; Yang, R.; Yang, Z.; Yao, S.; Zhao, S.; Wang, Y.; Li, P.; Song, X.; Jin, L.; Zhou, T.; et al. ROS Accumulation and Antiviral Defence Control by MicroRNA528 in Rice. Nat. Plants 2017, 3, 16203. [Google Scholar] [CrossRef]
- Urquiaga, M.C.d.O.; Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. From Trash to Luxury: The Potential Role of Plant LncRNA in DNA Methylation During Abiotic Stress. Front. Plant Sci. 2021, 11, 603246. [Google Scholar] [CrossRef]
- Zhao, Z.; Zang, S.; Zou, W.; Pan, Y.; Yao, W.; You, C. Long Non-Coding RNAs: New Players in Plants. Int. J. Mol. Sci 2022, 23, 9301. [Google Scholar] [CrossRef]
- Mortimer, J.C.; Yu, X.; Albrecht, S.; Sicilia, F.; Huichalaf, M.; Ampuero, D.; Michaelson, L.V.; Murphy, A.M.; Matsunaga, T.; Kurz, S.; et al. Abnormal Glycosphingolipid Mannosylation Triggers Salicylic Acid-Mediated Responses in Arabidopsis. Plant Cell 2013, 25, 1881–1894. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X.; Tangchaiburana, S.; Ndeh, R.; Markham, J.E.; Tsegaye, Y.; Dunn, T.M.; Wang, G.L.; Bellizzi, M.; Parsons, J.F.; et al. An Inositolphosphorylceramide Synthase Is Involved in Regulation of Plant Programmed Cell Death Associated with Defense in Arabidopsis. Plant Cell 2008, 20, 3163–3179. [Google Scholar] [CrossRef]
- Liebminger, E.; Veit, C.; Pabst, M.; Batoux, M.; Zipfel, C.; Altmann, F.; Mach, L.; Strasser, R. β-N-Acetylhexosaminidases HEXO1 and HEXO3 Are Responsible for the Formation of Paucimannosidic N-Glycans in Arabidopsis Thaliana. J. Biol. Chem. 2011, 286, 10793–10802. [Google Scholar] [CrossRef]
- Lannoo, N.; Van Damme, E.J.M. Review/N-Glycans: The Making of a Varied Toolbox. Plant Sci. 2015, 239, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Strasser, R. Plant Protein Glycosylation. Glycobiology 2016, 26, 926–939. [Google Scholar]
- Häweker, H.; Rips, S.; Koiwa, H.; Salomon, S.; Saijo, Y.; Chinchilla, D.; Robatzek, S.; Von Schaewen, A. Pattern Recognition Receptors Require N-Glycosylation to Mediate Plant Immunity. J. Biol. Chem. 2010, 285, 4629–4636. [Google Scholar] [CrossRef]
- Tör, M.; Gordon, P.; Cuzick, A.; Eulgem, T.; Sinapidou, E.; Mert-Türk, F.; Can, C.; Dangl, J.L.; Holub, E.B. Arabidopsis SGT1b Is Required for Defense Signaling Conferred by Several Downy Mildew Resistance Genes. Plant Cell 2002, 14, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.J.; Muskett, P.; Kahn, K.; Feys, B.J.; Jones, J.D.G.; Parker, J.E. Regulatory Role of SGT1 in Early R Gene-Mediated Plant Defenses. Science 2002, 295, 2077–2080. [Google Scholar]
- Fan, J.; Quan, W.; Li, G.B.; Hu, X.H.; Wang, Q.; Wang, H.; Li, X.P.; Luo, X.; Feng, Q.; Hu, Z.J.; et al. CircRNAs Are Involved in the Rice-Magnaporthe Oryzae Interaction. Plant Physiol. 2020, 182, 272–286. [Google Scholar] [CrossRef]
- Kong, K.W.; Mat-Junit, S.; Aminudin, N.; Ismail, A.; Abdul-Aziz, A. Antioxidant Activities and Polyphenolics from the Shoots of Barringtonia racemosa (L.) Spreng in a Polar to Apolar Medium System. Food Chem. 2012, 134, 324–332. [Google Scholar] [CrossRef]
- Gan, J.; Feng, Y.; He, Z.; Li, X.; Zhang, H. Correlations between Antioxidant Activity and Alkaloids and Phenols of Maca (Lepidium meyenii). J. Food Qual. 2017, 2017, 3185945. [Google Scholar] [CrossRef]
- Singh, R.; Singh, S.; Kumar, S.; Arora, S. Evaluation of Antioxidant Potential of Ethyl Acetate Extract/Fractions of Acacia Auriculiformis A. Cunn. Food Chem. Toxicol. 2007, 45, 1216–1223. [Google Scholar] [CrossRef]
- Gao, T.; Zhang, Y.; Shi, J.; Mohamed, S.R.; Xu, J.; Liu, X. The Antioxidant Guaiacol Exerts Fungicidal Activity Against Fungal Growth and Deoxynivalenol Production in Fusarium Graminearum. Front. Microbiol. 2021, 12, 762844. [Google Scholar] [CrossRef]
- Mueed, A.; Shibli, S.; Al-Quwaie, D.A.; Ashkan, M.F.; Alharbi, M.; Alanazi, H.; Binothman, N.; Aljadani, M.; Majrashi, K.A.; Huwaikem, M.; et al. Extraction, Characterization of Polyphenols from Certain Medicinal Plants and Evaluation of Their Antioxidant, Antitumor, Antidiabetic, Antimicrobial Properties, and Potential Use in Human Nutrition. Front. Nutr. 2023, 10, 1125106. [Google Scholar] [CrossRef]
- Yoshida, S.; Tamaoki, M.; Shikano, T.; Nakajima, N.; Ogawa, D.; Ioki, M.; Aono, M.; Kubo, A.; Kamada, H.; Inoue, Y.; et al. Cytosolic Dehydroascorbate Reductase Is Important for Ozone Tolerance in Arabidopsis Thaliana. Plant Cell Physiol. 2006, 47, 304–308. [Google Scholar] [CrossRef]
- Dicko, M.H.; Gruppen, H.; Hilhorst, R.; Voragen, A.G.J.; Van Berkel, W.J.H. Biochemical Characterization of the Major Sorghum Grain Peroxidase. FEBS J. 2006, 273, 2293–2307. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Hatan, E.; Kumar, V.; Galsurker, O.; Nisim-Levi, A.; Ovadia, R.; Galili, G.; Lewinsohn, E.; Elad, Y.; Alkan, N.; et al. Increased Phenylalanine Levels in Plant Leaves Reduces Susceptibility to Botrytis Cinerea. Plant Sci. 2020, 290, 110289. [Google Scholar] [CrossRef]
- Wang, X.; Che, M.Z.; Khalil, H.B.; McCallum, B.D.; Bakkeren, G.; Rampitsch, C.; Saville, B.J. The Role of Reactive Oxygen Species in the Virulence of Wheat Leaf Rust Fungus Puccinia Triticina. Environ. Microbiol. 2020, 22, 2956–2967. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Percaccio, E.; Gullì, M.; Romano, A.; Vitalone, A.; Mazzanti, G.; Gaetani, S.; Di Sotto, A. Recent Advances in the Neuroprotective Properties of Ferulic Acid in Alzheimer’s Disease: A Narrative Review. Nutrients 2022, 14, 3709. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Samota, M.K.; Choudhary, M.; Choudhary, M.; Pandey, A.K.; Sharma, A.; Thakur, J. How Do Plants Defend Themselves against Pathogens-Biochemical Mechanisms and Genetic Interventions. Physiol. Mol. Biol. Plants 2022, 28, 485–504. [Google Scholar] [CrossRef]
- Liu, Y.H.; Song, Y.H.; Ruan, Y.L. Sugar Conundrum in Plant-Pathogen Interactions: Roles of Invertase and Sugar Transporters Depend on Pathosystems. J. Exp. Bot. 2022, 73, 1910–1925. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.; Liu, Y.; Li, X.; Hao, G.; Han, Q.; Dirk, L.M.A.; Downie, A.B.; Ruan, Y.; Wang, J.; et al. Raffinose Synthase Enhances Drought Tolerance through Raffinose Synthesis or Galactinol Hydrolysis in Maize and Arabidopsis Plants. J. Biol. Chem. 2020, 295, 8064–8077. [Google Scholar] [CrossRef]
- Sood, S.G.; Comstock, J.C.; Glynn, N.C. Leaf Whorl Inoculation Method for Screening Sugarcane Rust Resistance. Plant Dis. 2009, 93, 1335–1340. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map (SAM) Format and SAMtools 1000 Genome Project Data Processing Subgroup. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A Tool for Long Non-Coding RNA Annotation and Its Application to the Dog Transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Kuang, Z.; Wang, Y.; Li, L.; Yang, X. MiRDeep-P2: Accurate and Fast Analysis of the MicroRNA Transcriptome in Plants. Bioinformatics 2019, 35, 2521–2522. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA Targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast Folding and Comparison of RNA Secondary Structures. Monatshefte Chem. Chem. Mon. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Zuker, M.; Stiegler, P. Optimal Computer Folding of Large RNA Sequences Using Thermodynamics and Auxiliary Information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef]
- McCaskill, J.S. The Equilibrium Partition Function and Base Pair Binding Probabilities for RNA Secondary Structure. Biopolymers 1990, 29, 1105–1119. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Schmid, R.; Heuckeroth, S.; Korf, A.; Smirnov, A.; Myers, O.; Dyrlund, T.S.; Bushuiev, R.; Murray, K.J.; Hoffmann, N.; Lu, M.; et al. Integrative Analysis of Multimodal Mass Spectrometry Data in MZmine 3. Nat. Biotechnol. 2023, 41, 447–449. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Induced Metabolite | Row Retention Time-Row m/z | Positive/Negative Mode | Line |
|---|---|---|---|
| 2,3-dihydroxyisovalerate | 133.0507-1.38 | Negative Mode | 540 |
| 3-4-hydroxyphenylpyruvate | 163.0387-7.52 | Positive Mode | 540 |
| 3-hydroxyphenylacetate | 151.0400-8.30 | Negative Mode | 540 |
| 4-hydroxyphenylacetate/mandelic acid | 151.0400-7.11 | Negative Mode | 540 |
| 6-deoxy-l-galactose | 163.0611-0.96 | Negative Mode | 540 |
| allantoin | 157.0367-0.81 | Negative Mode | 540 |
| 193.0135-0.80 | Negative Mode | 540 | |
| arabinose | 195.0509-0.73 | Negative Mode | 540 |
| D—galacturonic acid | 175.0236-0.89 | Negative Mode | 540 |
| D-glyceric acid | 233.0295-0.90 | Negative Mode | 540 |
| D—raffinose | 503.1617-0.93 | Negative Mode | 540 |
| 549.1674-0.96 | Negative Mode | 540 | |
| ferulate | 193.0500-9.32 | Negative Mode | 540 |
| glucose/fructose | 215.0328-0.74 | Negative Mode | 540 |
| 217.0296-0.74 | Negative Mode | 540 | |
| ll-2,6-diaminoheptanedioate | 189.0881-0.83 | Negative Mode | 540 |
| orotate | 155.0099-1.25 | Negative Mode | 540 |
| phenethylamine | 105.0702-7.23 | Positive Mode | 540 |
| theobromine | 181.0729-7.00 | Positive Mode | 540 |
| uracil | 111.0199-1.25 | Negative Mode | 540 |
| 11-dehydro-2,3-dinor TXB2 | 339.1809-10.33 | Negative Mode | 664 |
| 3-hydroxy-3-methylglutarate | 161.0455-3.13 | Negative Mode | 664 |
| 183.0274-3.15 | Negative Mode | 664 | |
| 185.0418-3.10 | Positive Mode | 664 | |
| betaine | 152.0481-0.78 | Negative Mode | 664 |
| l-arginine | 209.0800-0.74 | Negative Mode | 664 |
| 2-acetamido-2-deoxy-beta-D-glucosylamine | 203.1025-0.88 | Positive Mode | 540 664 |
| 2-Hydroxyhippuric acid | 194.0459-7.57 | Negative Mode | 540 664 |
| caffeate | 163.0388-8.31 | Positive Mode | 540 664 |
| catechol | 109.0291-7.24 | Negative Mode | 540 664 |
| phenylpyruvate | 209.0454-8.42 | Negative Mode | 540 664 |
| 209.0459-8.33 | Negative Mode | 540 664 | |
| pyridoxine | 170.0808-2.94 | Positive Mode | 540 664 |
| shikimate | 219.0510-0.95 | Negative Mode | 664 |
| 219.0510-1.13 | Negative Mode | 540 664 | |
| 219.0510-1.20 | Negative Mode | 540 664 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Mihail, E.S.; Luo, Z.; Sood, S.; Islam, M.S.; Wang, J. Exploring Morphological, Transcriptomic, and Metabolomic Differences Between Two Sister Lines with Contrasting Resistance to Orange Rust Disease in Sugarcane. Int. J. Mol. Sci. 2025, 26, 3490. https://doi.org/10.3390/ijms26083490
Zhou Y, Mihail ES, Luo Z, Sood S, Islam MS, Wang J. Exploring Morphological, Transcriptomic, and Metabolomic Differences Between Two Sister Lines with Contrasting Resistance to Orange Rust Disease in Sugarcane. International Journal of Molecular Sciences. 2025; 26(8):3490. https://doi.org/10.3390/ijms26083490
Chicago/Turabian StyleZhou, Yupeng, Edvin Sebastian Mihail, Ziliang Luo, Sushma Sood, Md Sariful Islam, and Jianping Wang. 2025. "Exploring Morphological, Transcriptomic, and Metabolomic Differences Between Two Sister Lines with Contrasting Resistance to Orange Rust Disease in Sugarcane" International Journal of Molecular Sciences 26, no. 8: 3490. https://doi.org/10.3390/ijms26083490
APA StyleZhou, Y., Mihail, E. S., Luo, Z., Sood, S., Islam, M. S., & Wang, J. (2025). Exploring Morphological, Transcriptomic, and Metabolomic Differences Between Two Sister Lines with Contrasting Resistance to Orange Rust Disease in Sugarcane. International Journal of Molecular Sciences, 26(8), 3490. https://doi.org/10.3390/ijms26083490