
BRAF Targeting Across Solid Tumors: Molecular Aspects and Clinical Applications

 , ,
, ,
Abstract
1. Introduction
2. BRAF Biology and Its Therapeutical Implication
2.1. Impact of BRAF Mutation on Prognosis
2.2. Implication of BRAF Mutation on Immunotherapy Efficacy
3. Selected Strategies in BRAF-Mutated Solid Cancers
3.1. Melanoma
3.2. Advanced Non-Small-Cell Lung Cancer
3.3. Thyroid Cancer
3.4. Metastatic Colorectal Cancer
4. Agnostic Approach to BRAF Inhibition: Selected Examples of Rare Tumors
5. Mechanisms of Resistance
5.1. Intrinsic Resistance
5.2. Acquired Resistance
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf Kinases: Function, Regulation and Role in Human Cancer. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, A.J.; Chen, Z.; Rosen, N.; Solit, D.B. BRAF—A Tumour-Agnostic Drug Target with Lineage-Specific Dependencies. Nat. Rev. Clin. Oncol. 2024, 21, 224–247. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Desideri, E.; Cavallo, A.L.; Baccarini, M. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 2015, 161, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential Regulation and Properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of Receptor Tyrosine Kinase Activation in Cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Dibb, N.J.; Dilworth, S.M.; Mol, C.D. Switching on Kinases: Oncogenic Activation of BRAF and the PDGFR Family. Nat. Rev. Cancer 2004, 4, 718–727. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF–MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing Clinicopathologic Features of Patients with V600E and V600K BRAF-Mutant Metastatic Melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.L.; Vidwans, S.J.; Janku, F.; Garrido-Laguna, I.; Munoz, J.; Schwab, R.; Subbiah, V.; Rodon, J.; Kurzrock, R. Genomically Driven Tumors and Actionability across Histologies: BRAF-Mutant Cancers as a Paradigm. Mol. Cancer Ther. 2016, 15, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with Class 3 BRAF Mutants Are Sensitive to the Inhibition of Activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Frisone, D.; Friedlaender, A.; Malapelle, U.; Banna, G.; Addeo, A. A BRAF New World. Crit. Rev. Oncol./Hematol. 2020, 152, 103008. [Google Scholar] [CrossRef]
- Özgü, E.; Kaplan, B.G.; Sivakumar, S.; Sokol, E.S.; Aydın, E.; Tokat, Ü.M.; Adibi, A.; Karakoç, E.G.; Hu, J.; Kurzrock, R.; et al. Therapeutic Vulnerabilities and Pan-Cancer Landscape of BRAF Class III Mutations in Epithelial Solid Tumors. BJC Rep. 2024, 2, 77. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus Trametinib in Patients with Previously Untreated BRAFV600E-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Gazzah, A.; Lassen, U.; Stein, A.; Wen, P.Y.; Dietrich, S.; De Jonge, M.J.A.; Blay, J.-Y.; et al. Dabrafenib plus Trametinib in BRAFV600E-Mutated Rare Cancers: The Phase 2 ROAR Trial. Nat. Med. 2023, 29, 1103–1112. [Google Scholar] [CrossRef]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association Between NRAS and BRAF Mutational Status and Melanoma-Specific Survival Among Patients with Higher-Risk Primary Melanoma. JAMA Oncol. 2015, 1, 359. [Google Scholar] [CrossRef]
- Pires Da Silva, I.; Wang, K.Y.X.; Wilmott, J.S.; Holst, J.; Carlino, M.S.; Park, J.J.; Quek, C.; Wongchenko, M.; Yan, Y.; Mann, G.; et al. Distinct Molecular Profiles and Immunotherapy Treatment Outcomes of V600E and V600K BRAF-Mutant Melanoma. Clin. Cancer Res. 2019, 25, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.P.; Tang, Y.H.; Armour, N.; Dutton-Regester, K.; Krause, L.; Loffler, K.A.; Lambie, D.; Burmeister, B.; Thomas, J.; Smithers, B.M.; et al. BRAF Mutation Status Is an Independent Prognostic Factor for Resected Stage IIIB and IIIC Melanoma: Implications for Melanoma Staging and Adjuvant Therapy. Eur. J. Cancer 2014, 50, 2668–2676. [Google Scholar] [CrossRef]
- Clarke, C.N.; Kopetz, E.S. BRAF Mutant Colorectal Cancer as a Distinct Subset of Colorectal Cancer: Clinical Characteristics, Clinical Behavior, and Response to Targeted Therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, E.; Bennett, M.W.; Daly, L.; Power, D.G. The Implication of BRAF Mutation in Advanced Colorectal Cancer. Ir. J. Med. Sci. 2022, 191, 2467–2474. [Google Scholar] [CrossRef]
- Thibodeau, S.N.; French, A.J.; Cunningham, J.M.; Tester, D.; Burgart, L.J.; Roche, P.C.; McDonnell, S.K.; Schaid, D.J.; Vockley, C.W.; Michels, V.V.; et al. Microsatellite Instability in Colorectal Cancer: Different Mutator Phenotypes and the Principal Involvement of hMLH1. Cancer Res. 1998, 58, 1713–1718. [Google Scholar]
- Bond, C.E.; Whitehall, V.L.J. How the BRAF V600E Mutation Defines a Distinct Subgroup of Colorectal Cancer: Molecular and Clinical Implications. Gastroenterol. Res. Pract. 2018, 2018, 9250757. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus Bevacizumab versus FOLFIRI plus Bevacizumab as First-Line Treatment of Patients with Metastatic Colorectal Cancer: Updated Overall Survival and Molecular Subgroup Analyses of the Open-Label, Phase 3 TRIBE Study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T.R.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Poor Survival Associated with the BRAF V600E Mutation in Microsatellite-Stable Colon Cancers. Cancer Res. 2005, 65, 6063–6069. [Google Scholar] [CrossRef]
- Lochhead, P.; Kuchiba, A.; Imamura, Y.; Liao, X.; Yamauchi, M.; Nishihara, R.; Qian, Z.R.; Morikawa, T.; Shen, J.; Meyerhardt, J.A.; et al. Microsatellite Instability and BRAF Mutation Testing in Colorectal Cancer Prognostication. J. Natl. Cancer Inst. 2013, 105, 1151–1156. [Google Scholar] [CrossRef]
- Taieb, J.; Le Malicot, K.; Shi, Q.; Penault Lorca, F.; Bouché, O.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Folprecht, G.; Luc Van Laethem, J.; et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J. Natl. Cancer Inst. 2017, 109, djw272. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of Colorectal Cancer: Challenges for Therapeutic Efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.E.; Umapathy, A.; Buttenshaw, R.L.; Wockner, L.; Leggett, B.A.; Whitehall, V.L.J. Chromosomal Instability in BRAF Mutant, Microsatellite Stable Colorectal Cancers. PLoS ONE 2012, 7, e47483. [Google Scholar] [CrossRef]
- Rizzo, A.; Federico, A.D.; Ricci, A.D.; Frega, G.; Palloni, A.; Pagani, R.; Tavolari, S.; Marco, M.D.; Brandi, G. Targeting BRAF-Mutant Biliary Tract Cancer: Recent Advances and Future Challenges. Cancer Control 2020, 27, 107327482098301. [Google Scholar] [CrossRef]
- Li, W.; Cui, Y.; Yin, F.; Peng, L.; Liu, X.; Shen, Y.; Guo, Y.; Wen, S.; Shi, J.; Lei, M.; et al. BRAF Mutation in Chinese Biliary Tract Cancer Patients. J. Clin. Oncol. 2020, 38 (Suppl. S15), e16678. [Google Scholar] [CrossRef]
- Tannapfel, A. Mutations of the BRAF Gene in Cholangiocarcinoma but Not in Hepatocellular Carcinoma. Gut 2003, 52, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Hyder, O.; Dodson, R.; Nayar, S.K.; Poling, J.; Beierl, K.; Eshleman, J.R.; Lin, M.-T.; Pawlik, T.M.; Anders, R.A. The Frequency of KRAS and BRAF Mutations in Intrahepatic Cholangiocarcinomas and Their Correlation with Clinical Outcome. Hum. Pathol. 2013, 44, 2768–2773. [Google Scholar] [CrossRef]
- Marchetti, A.; Felicioni, L.; Malatesta, S.; Grazia Sciarrotta, M.; Guetti, L.; Chella, A.; Viola, P.; Pullara, C.; Mucilli, F.; Buttitta, F. Clinical Features and Outcome of Patients with Non–Small-Cell Lung Cancer Harboring BRAF Mutations. J. Clin. Oncol. 2011, 29, 3574–3579. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Banna, G.; Malapelle, U.; Pisapia, P.; Addeo, A. Next Generation Sequencing and Genetic Alterations in Squamous Cell Lung Carcinoma: Where Are We Today? Front. Oncol. 2019, 9, 166. [Google Scholar] [CrossRef]
- Wu, H.; Feng, J.; Lu, S.; Huang, J. A Large-Scale, Multicenter Characterization of BRAF G469V/A-Mutant Non-Small Cell Lung Cancer. Cancer Med. 2024, 13, e7305. [Google Scholar] [CrossRef]
- Su, P.-L.; Lin, C.-Y.; Chen, Y.-L.; Chen, W.-L.; Lin, C.-C.; Su, W.-C. Durable Response to Combined Dabrafenib and Trametinib in a Patient with BRAF K601E Mutation-Positive Lung Adenocarcinoma: A Case Report. JTO Clin. Res. Rep. 2021, 2, 100202. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Tseng, L.-H.; Chen, G.; Haley, L.; Illei, P.; Gocke, C.D.; Eshleman, J.R.; Lin, M.-T. Clinical Detection and Categorization of Uncommon and Concomitant Mutations Involving BRAF. BMC Cancer 2015, 15, 779. [Google Scholar] [CrossRef] [PubMed]
- Faehling, M.; Schwenk, B.; Kramberg, S.; Eckert, R.; Volckmar, A.-L.; Stenzinger, A.; Sträter, J. Oncogenic Driver Mutations, Treatment, and EGFR-TKI Resistance in a Caucasian Population with Non-Small Cell Lung Cancer: Survival in Clinical Practice. Oncotarget 2017, 8, 77897–77914. [Google Scholar] [CrossRef]
- Anguera, G.; Majem, M. BRAF Inhibitors in Metastatic Non-Small Cell Lung Cancer. J. Thorac. Dis. 2018, 10, 589–592. [Google Scholar] [CrossRef]
- Xing, M. BRAF Mutation in Thyroid Cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef]
- Choi, S.; Shugard, E.; Khanafshar, E.; Quivey, J.M.; Garsa, A.A.; Yom, S.S. Association Between BRAF V600E Mutation and Decreased Survival in Patients Locoregionally Irradiated for Anaplastic Thyroid Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, E356. [Google Scholar] [CrossRef]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M.; et al. Association Between BRAF V600E Mutation and Mortality in Patients with Papillary Thyroid Cancer. JAMA 2013, 309, 1493. [Google Scholar] [CrossRef]
- Tufano, R.P.; Teixeira, G.V.; Bishop, J.; Carson, K.A.; Xing, M. BRAF Mutation in Papillary Thyroid Cancer and Its Value in Tailoring Initial Treatment: A Systematic Review and Meta-Analysis. Medicine 2012, 91, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hu, S.; Hou, P.; Jiang, D.; Condouris, S.; Xing, M. Suppression of BRAF/MEK/MAP Kinase Pathway Restores Expression of Iodide-Metabolizing Genes in Thyroid Cells Expressing the V600E BRAF Mutant. Clin. Cancer Res. 2007, 13, 1341–1349. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, D. Synergistic Inhibition of MEK/ERK and BRAF V600E with PD98059 and PLX4032 Induces Sodium/Iodide Symporter (NIS) Expression and Radioiodine Uptake in BRAF Mutated Papillary Thyroid Cancer Cells. Thyroid. Res. 2018, 11, 13. [Google Scholar] [CrossRef]
- Hou, P.; Bojdani, E.; Xing, M. Induction of Thyroid Gene Expression and Radioiodine Uptake in Thyroid Cancer Cells by Targeting Major Signaling Pathways. J. Clin. Endocrinol. Metab. 2010, 95, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Weichenthal, M.; Svane, I.M.; Kandolf Sekulovic, L.; Mangana, J.; Mohr, P.; Marquez-Rodas, I.; Schmidt, H.; Ziogas, D.C.; Bender, M.; Ellebaek, E.; et al. EMRseq: Registry-Based Outcome Analysis on 1,000 Patients with BRAF V600–Mutated Metastatic Melanoma in Europe Treated with Either Immune Checkpoint or BRAF-/MEK Inhibition. J. Clin. Oncol. 2022, 40 (Suppl. S16), 9540. [Google Scholar] [CrossRef]
- André, T.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Nivolumab plus Low-Dose Ipilimumab in Previously Treated Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: 4-Year Follow-up from CheckMate 142. Ann. Oncol. 2022, 33, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Mechahougui, H.; Friedlaender, A. Unraveling the Nexus: Oncogenic Drivers and Immunotherapy Efficacy in Cancer Treatment. Immunotherapy 2024, 16, 267–271. [Google Scholar] [CrossRef]
- Park, R.; Lopes, L.; Lee, S.; Riano, I.; Saeed, A. The Prognostic and Predictive Impact of BRAF Mutations in Deficient Mismatch Repair/Microsatellite Instability-High Colorectal Cancer: Systematic Review/Meta-Analysis. Future Oncol. 2021, 17, 4221–4231. [Google Scholar] [CrossRef]
- Nguyen-Ngoc, T.; Bouchaab, H.; Adjei, A.A.; Peters, S. BRAF Alterations as Therapeutic Targets in Non–Small-Cell Lung Cancer. J. Thorac. Oncol. 2015, 10, 1396–1403. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Xu, Y.; Huang, Z.; Cheng, G.; Xie, M.; Zhou, Z.; Yu, Y.; Xi, W.; Fan, Y. Tumor Immune Microenvironment and Immunotherapy Efficacy in BRAF Mutation Non-Small-Cell Lung Cancer. Cell Death Dis. 2022, 13, 1064. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Oncogene-Addicted Metastatic Non-Small-Cell Lung Cancer: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2023, 34, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Comin-Anduix, B.; Chodon, T.; Sazegar, H.; Matsunaga, D.; Mock, S.; Jalil, J.; Escuin-Ordinas, H.; Chmielowski, B.; Koya, R.C.; Ribas, A. The Oncogenic BRAF Kinase Inhibitor PLX4032/RG7204 Does Not Affect the Viability or Function of Human Lymphocytes across a Wide Range of Concentrations. Clin. Cancer Res. 2010, 16, 6040–6048. [Google Scholar] [CrossRef]
- Holderfield, M.; Nagel, T.E.; Stuart, D.D. Mechanism and Consequences of RAF Kinase Activation by Small-Molecule Inhibitors. Br. J. Cancer 2014, 111, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Vence, L.; Falchook, G.; Radvanyi, L.G.; Liu, C.; Goodman, V.; Legos, J.J.; Blackman, S.; Scarmadio, A.; Kurzrock, R.; et al. BRAF(V600) Inhibitor GSK2118436 Targeted Inhibition of Mutant BRAF in Cancer Patients Does Not Impair Overall Immune Competency. Clin. Cancer Res. 2012, 18, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory Effects of BRAF and MEK Inhibitors: Implications for Melanoma Therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Simeone, E.; Sileni, V.C.; Del Vecchio, M.; Marchetti, P.; Cappellini, G.C.A.; Ridolfi, R.; de Rosa, F.; Cognetti, F.; Ferraresi, V.; et al. Sequential Treatment with Ipilimumab and BRAF Inhibitors in Patients with Metastatic Melanoma: Data from the Italian Cohort of the Ipilimumab Expanded Access Program. Cancer Investig. 2014, 32, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.-G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients with Advanced BRAF-Mutant Melanoma: The DREAMseq Trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and Trametinib versus Dabrafenib and Placebo for Val600 BRAF-Mutant Melanoma: A Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Cortot, A.B.; Audigier-Valette, C.; Molinier, O.; Le Moulec, S.; Barlesi, F.; Zalcman, G.; Dumont, P.; Pouessel, D.; Poulet, C.; Fontaine-Delaruelle, C.; et al. Weekly Paclitaxel plus Bevacizumab versus Docetaxel as Second- or Third-Line Treatment in Advanced Non-Squamous Non–Small-Cell Lung Cancer: Results of the IFCT-1103 ULTIMATE Study. Eur. J. Cancer 2020, 131, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular Testing for BRAF Mutations to Inform Melanoma Treatment Decisions: A Move toward Precision Medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers 2023, 15, 4026. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; McKee, A.E.; Ning, Y.-M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA Approval Summary: Vemurafenib for Treatment of Unresectable or Metastatic Melanoma with the BRAFV600E Mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in Patients with BRAFV600 Mutation-Positive Metastatic Melanoma: Final Overall Survival Results of the Randomized BRIM-3 Study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Maio, M.; Arance, A.; Nathan, P.; Blank, C.; Avril, M.-F.; Garbe, C.; Hauschild, A.; Schadendorf, D.; Hamid, O.; et al. Vemurafenib in Metastatic Melanoma Patients with Brain Metastases: An Open-Label, Single-Arm, Phase 2, Multicentre Study. Ann. Oncol. 2017, 28, 634–641. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. An Update on BREAK-3, a Phase III, Randomized Trial: Dabrafenib (DAB) versus Dacarbazine (DTIC) in Patients with BRAF V600E-Positive Mutation Metastatic Melanoma (MM). J. Clin. Oncol. 2013, 31 (Suppl. S15), 9013. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Flaherty, K.; Nathan, P.; Hersey, P.; Garbe, C.; Milhem, M.; Demidov, L.; Mohr, P.; Hassel, J.C.; Rutkowski, P.; et al. Five-Year Outcomes from a Phase 3 METRIC Study in Patients with BRAF V600 E/K–Mutant Advanced or Metastatic Melanoma. Eur. J. Cancer 2019, 109, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib Combined with Vemurafenib in Advanced BRAFV600-Mutant Melanoma (coBRIM): Updated Efficacy Results from a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Dréno, B.; Larkin, J.; Ribas, A.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. 5-Year Outcomes with Cobimetinib plus Vemurafenib in BRAF V600 Mutation–Positive Advanced Melanoma: Extended Follow-up of the coBRIM Study. Clin. Cancer Res. 2021, 27, 5225–5235. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovsky, D.L.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.M.G.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. COMBI-d: A Randomized, Double-Blinded, Phase III Study Comparing the Combination of Dabrafenib and Trametinib to Dabrafenib and Trametinib Placebo as First-Line Therapy in Patients (Pts) with Unresectable or Metastatic BRAF V600E/K Mutation-Positive Cutaneous Melanoma. J. Clin. Oncol. 2014, 32 (Suppl. S15), 9011. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus Trametinib versus Dabrafenib Monotherapy in Patients with Metastatic BRAF V600E/K-Mutant Melanoma: Long-Term Survival and Safety Analysis of a Phase 3 Study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.-J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L.; et al. Dabrafenib plus Trametinib in Patients with BRAFV600-Mutant Melanoma Brain Metastases (COMBI-MB): A Multicentre, Multicohort, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib in Patients with BRAF -Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall Survival in Patients with BRAF-Mutant Melanoma Receiving Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib (COLUMBUS): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; De Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsová, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients with BRAF V600–Mutant Melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Dudnik, E.; Peled, N.; Nechushtan, H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J. Thorac. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D. BRAF in Non-Small Cell Lung Cancer (NSCLC): Pickaxing Another Brick in the Wall. Cancer Treat. Rev. 2018, 66, 82–94. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Martinez, P.; Yeap, B.Y.; Ambrogio, C.; Ferris, L.A.; Lydon, C.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Impact of BRAF Mutation Class on Disease Characteristics and Clinical Outcomes in BRAF-Mutant Lung Cancer. Clin. Cancer Res. 2019, 25, 158–165. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non–Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef]
- Dankner, M.; Lajoie, M.; Moldoveanu, D.; Nguyen, T.-T.; Savage, P.; Rajkumar, S.; Huang, X.; Lvova, M.; Protopopov, A.; Vuzman, D.; et al. Dual MAPK Inhibition Is an Effective Therapeutic Strategy for a Subset of Class II BRAF Mutant Melanomas. Clin. Cancer Res. 2018, 24, 6483–6494. [Google Scholar] [CrossRef]
- Gautschi, O.; Milia, J.; Cabarrou, B.; Bluthgen, M.-V.; Besse, B.; Smit, E.F.; Wolf, J.; Peters, S.; Früh, M.; Koeberle, D.; et al. Targeted Therapy for Patients with BRAF-Mutant Lung Cancer Results from the European EURAF Cohort. J. Thorac. Oncol. 2015, 10, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gervais, R.; Riely, G.; Hollebecque, A.; Blay, J.-Y.; Felip, E.; Schuler, M.; Gonçalves, A.; Italiano, A.; Keedy, V.; et al. Efficacy of Vemurafenib in Patients with Non–Small-Cell Lung Cancer with BRAF V600 Mutation: An Open-Label, Single-Arm Cohort of the Histology-Independent VE-BASKET Study. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Montané, L.; Barlesi, F.; Coudert, B.; Souquet, P.J.; Otto, J.; Gervais, R.; Moro-Sibilot, D.; Monnet, I.; Brain, E.; et al. OA12.05 Vemurafenib in Patients Harboring V600 and Non V600 BRAF Mutations: Final Results of the NSCLC Cohort from the AcSé Trial. J. Thorac. Oncol. 2018, 13, S348–S349. [Google Scholar] [CrossRef]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in Non-Small-Cell Lung Cancer Patients with BRAFV600 and BRAFnonV600 Mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef]
- Reyes, R.; Mayo-de-las-Casas, C.; Teixidó, C.; Cabrera, C.; Marín, E.; Vollmer, I.; Jares, P.; Garzón, M.; Molina-Vila, M.Á.; Reguart, N. Clinical Benefit From BRAF/MEK Inhibition in a Double Non-V600E BRAF Mutant Lung Adenocarcinoma: A Case Report. Clin. Lung Cancer 2019, 20, e219–e223. [Google Scholar] [CrossRef]
- Kotani, H.; Adachi, Y.; Kitai, H.; Tomida, S.; Bando, H.; Faber, A.C.; Yoshino, T.; Voon, D.C.; Yano, S.; Ebi, H. Distinct Dependencies on Receptor Tyrosine Kinases in the Regulation of MAPK Signaling between BRAF V600E and Non-V600E Mutant Lung Cancers. Oncogene 2018, 37, 1775–1787. [Google Scholar] [CrossRef]
- Sereno, M.; Moreno, V.; Moreno Rubio, J.; Gómez-Raposo, C.; García Sánchez, S.; Hernández Jusdado, R.; Falagan, S.; Zambrana Tébar, F.; Casado Sáenz, E. A Significant Response to Sorafenib in a Woman with Advanced Lung Adenocarcinoma and a BRAF Non-V600 Mutation. Anti-Cancer Drugs 2015, 26, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Casadei Gardini, A.; Chiadini, E.; Faloppi, L.; Marisi, G.; Delmonte, A.; Scartozzi, M.; Loretelli, C.; Lucchesi, A.; Oboldi, D.; Dubini, A.; et al. Efficacy of Sorafenib in BRAF-Mutated Non-Small-Cell Lung Cancer (NSCLC) and No Response in Synchronous BRAF Wild Type-Hepatocellular Carcinoma: A Case Report. BMC Cancer 2016, 16, 429. [Google Scholar] [CrossRef]
- Clark, J.W.; Eder, J.P.; Ryan, D.; Lathia, C.; Lenz, H.-J. Safety and Pharmacokinetics of the Dual Action Raf Kinase and Vascular Endothelial Growth Factor Receptor Inhibitor, BAY 43-9006, in Patients with Advanced, Refractory Solid Tumors. Clin. Cancer Res. 2005, 11, 5472–5480. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Pan, J.; Zhou, H.; Zhu, S.; Huang, J.; Zhao, X.; Ding, H.; Pan, Y. Development of Small-Molecule Therapeutics and Strategies for Targeting RAF Kinase in BRAF-Mutant Colorectal Cancer. Cancer Manag. Res. 2018, 10, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Tutuka, C.S.A.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a New Generation BRAF Inhibitor, Selectively Inhibits BRAF in Colonic Adenocarcinoma Cells and Prevents Paradoxical MAPK Pathway Activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Hollebecque, A.; Flaherty, K.T.; Shapiro, G.I.; Rodon Ahnert, J.; Millward, M.J.; Zhang, W.; Gao, L.; Sykes, A.; Willard, M.D.; et al. A Phase I Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol. Cancer Ther. 2020, 19, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, J.; Shin, S.J.; Kim, J.-S.; Kim, Y.J.; Han, H.S.; Lee, S.J.; Lim, H.-S.; Hong, Y.; Noh, Y.S.; et al. Belvarafenib, a Novel Pan-RAF Inhibitor, in Solid Tumor Patients Harboring BRAF, KRAS, or NRAS Mutations: Phase I Study. J. Clin. Oncol. 2019, 37 (Suppl. S15), 3000. [Google Scholar] [CrossRef]
- Leboulleux, S.; Benisvy, D.; Taieb, D.; Attard, M.; Bournaud, C.; Terroir, M.; Al Ghuzlan, A.; Lamartina, L.; Schlumberger, M.J.; Godbert, Y.; et al. 1743MO MERAIODE: A Redifferentiation Phase II Trial with Trametinib Followed by Radioactive Iodine for Metastatic Radioactive Iodine Refractory Differentiated Thyroid Cancer Patients with a RAS Mutation. Ann. Oncol. 2021, 32, S1204. [Google Scholar] [CrossRef]
- Leboulleux, S.; Benisvy, D.; Taieb, D.; Attard, M.; Bournaud, C.; Terroir-Cassou-Mounat, M.; Lacroix, L.; Anizan, N.; Schiazza, A.; Garcia, M.E.; et al. MERAIODE: A Phase II Redifferentiation Trial with Trametinib and 131 I in Metastatic Radioactive Iodine Refractory RAS Mutated Differentiated Thyroid Cancer. Thyroid 2023, 33, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus Trametinib in Patients with BRAF V600E-Mutant Anaplastic Thyroid Cancer: Updated Analysis from the Phase II ROAR Basket Study. Ann. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.W.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in Patients with BRAFV600E-Positive Metastatic or Unresectable Papillary Thyroid Cancer Refractory to Radioactive Iodine: A Non-Randomised, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Chu, J.E.; Johnson, B.; Kugathasan, L.; Morris, V.K.; Raghav, K.; Swanson, L.; Lim, H.J.; Renouf, D.J.; Gill, S.; Wolber, R.; et al. Population-Based Screening for BRAF V600E in Metastatic Colorectal Cancer Reveals Increased Prevalence and Poor Prognosis. Clin. Cancer Res. 2020, 26, 4599–4605. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, E.; Ristimäki, A.; Mäkinen, M.J.; Kytölä, S.; Kononen, J.; Pfeiffer, P.; Soveri, L.; Keinänen, M.; Sorbye, H.; Nunes, L.; et al. Atypical (non-V600E) BRAF Mutations in Metastatic Colorectal Cancer in Population and Real-world Cohorts. Int. J. Cancer 2024, 154, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.M.; Esmail, A.; Abdelrahim, M. Triple-Regimen of Vemurafenib, Irinotecan, and Cetuximab for the Treatment of BRAFV600E-Mutant CRC: A Case Report and Review. Front. Pharmacol. 2021, 12, 795381. [Google Scholar] [CrossRef]
- Vranic, S.; Basu, G.D.; Hall, D.W.; Gatalica, Z. Tumor-Type Agnostic, Targeted Therapies: BRAF Inhibitors Join the Group. Acta Medica Acad. 2023, 51, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Edeline, J.; Goyal, L. How I Treat Biliary Tract Cancer. ESMO Open 2022, 7, 100378. [Google Scholar] [CrossRef]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.-I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients with Tumors with BRAF V600E Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Di Nunno, V.; Gatto, L.; Tosoni, A.; Bartolini, S.; Franceschi, E. Implications of BRAF V600E Mutation in Gliomas: Molecular Considerations, Prognostic Value and Treatment Evolution. Front. Oncol. 2023, 12, 1067252. [Google Scholar] [CrossRef]
- Szklener, K.; Mazurek, M.; Wieteska, M.; Wacławska, M.; Bilski, M.; Mańdziuk, S. New Directions in the Therapy of Glioblastoma. Cancers 2022, 14, 5377. [Google Scholar] [CrossRef]
- Mittapalli, R.K.; Vaidhyanathan, S.; Dudek, A.Z.; Elmquist, W.F. Mechanisms Limiting Distribution of the Threonine-Protein Kinase B-RaFV600E Inhibitor Dabrafenib to the Brain: Implications for the Treatment of Melanoma Brain Metastases. J. Pharmacol. Exp. Ther. 2013, 344, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Carter, T.; Kitchen, N.; Mulholland, P. Dabrafenib and Trametinib in BRAFV600E Mutated Glioma. CNS Oncol. 2017, 6, 291–296. [Google Scholar] [CrossRef]
- Lim-Fat, M.J.; Song, K.W.; Iorgulescu, J.B.; Andersen, B.M.; Forst, D.A.; Jordan, J.T.; Gerstner, E.R.; Reardon, D.A.; Wen, P.Y.; Arrillaga-Romany, I. Clinical, Radiological and Genomic Features and Targeted Therapy in BRAF V600E Mutant Adult Glioblastoma. J. Neurooncol 2021, 152, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.-D.; Joly, F.; et al. BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study. J. Clin. Oncol. 2018, 36, 3477–3484. [Google Scholar] [CrossRef]
- Gadducci, A.; Cosio, S. Therapeutic Approach to Low-Grade Serous Ovarian Carcinoma: State of Art and Perspectives of Clinical Research. Cancers 2020, 12, 1336. [Google Scholar] [CrossRef] [PubMed]
- Zwimpfer, T.A.; Tal, O.; Geissler, F.; Coelho, R.; Rimmer, N.; Jacob, F.; Heinzelmann-Schwarz, V. Low Grade Serous Ovarian Cancer—A Rare Disease with Increasing Therapeutic Options. Cancer Treat. Rev. 2023, 112, 102497. [Google Scholar] [CrossRef]
- Pugh, T.J.; Bell, J.L.; Bruce, J.P.; Doherty, G.J.; Galvin, M.; Green, M.F.; Hunter-Zinck, H.; Kumari, P.; Lenoue-Newton, M.L.; Li, M.M.; et al. AACR Project GENIE: 100,000 Cases and Beyond. Cancer Discov. 2022, 12, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Grisham, R.N.; Banerjee, S.; Kalbacher, E.; Mirza, M.R.; Romero, I.; Vuylsteke, P.; Coleman, R.L.; Hilpert, F.; Oza, A.M.; et al. MILO/ENGOT-Ov11: Binimetinib Versus Physician’s Choice Chemotherapy in Recurrent or Persistent Low-Grade Serous Carcinomas of the Ovary, Fallopian Tube, or Primary Peritoneum. J. Clin. Oncol. 2020, 38, 3753–3762. [Google Scholar] [CrossRef]
- Grisham, R.N.; Vergote, I.; Banerjee, S.; Drill, E.; Kalbacher, E.; Mirza, M.R.; Romero, I.; Vuylsteke, P.; Coleman, R.L.; Hilpert, F.; et al. Molecular Results and Potential Biomarkers Identified from the Phase 3 MILO/ENGOT-Ov11 Study of Binimetinib versus Physician Choice of Chemotherapy in Recurrent Low-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2023, 29, 4068–4075. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Miller, A.; Brady, W.E.; Paul, J.; Carty, K.; Rodgers, W.; Millan, D.; Coleman, R.L.; Moore, K.N.; Banerjee, S.; et al. Trametinib versus Standard of Care in Patients with Recurrent Low-Grade Serous Ovarian Cancer (GOG 281/LOGS): An International, Randomised, Open-Label, Multicentre, Phase 2/3 Trial. Lancet 2022, 399, 541–553. [Google Scholar] [CrossRef]
- Mechahougui, H.; Michael, M.; Friedlaender, A. Precision Oncology in Gastrointestinal Stromal Tumors. Curr. Oncol. 2023, 30, 4648–4662. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Furney, S.J.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-Genome Sequencing Reveals Complex Mechanisms of Intrinsic Resistance to BRAF Inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.M.; Lioni, M.; Palma, M.D.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; Van Belle, P.; et al. Increased Cyclin D1 Expression Can Mediate BRAF Inhibitor Resistance in BRAF V600E–Mutated Melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Theurillat, J.-P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A Genome-Scale RNA Interference Screen Implicates NF1 Loss in Resistance to RAF Inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo Effector YAP Promotes Resistance to RAF- and MEK-Targeted Cancer Therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Vellano, C.P.; White, M.G.; Andrews, M.C.; Chelvanambi, M.; Witt, R.G.; Daniele, J.R.; Titus, M.; McQuade, J.L.; Conforti, F.; Burton, E.M.; et al. Androgen Receptor Blockade Promotes Response to BRAF/MEK-Targeted Therapy. Nature 2022, 606, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of Resistance to BRAF and MEK Inhibitors and Clinical Update of US Food and Drug Administration-Approved Targeted Therapy in Advanced Melanoma. Onco Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Martin, A.-M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.-T.; et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garrett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitner, D.; Santinami, M.; Atkinson, V.; Mandalà, M.; et al. Adjuvant Dabrafenib plus Trametinib versus Placebo in Patients with Resected, BRAFV600-Mutant, Stage III Melanoma (COMBI-AD): Exploratory Biomarker Analyses from a Randomised, Phase 3 Trial. Lancet Oncol. 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Fulton-Ward, T.; Middleton, G. The Impact of Genomic Context on Outcomes of Solid Cancer Patients Treated with Genotype-Matched Targeted Therapies: A Comprehensive Review. Ann. Oncol. 2023, 34, 1113–1130. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, I.Z.M.; Slieker, R.C.; Van Groningen, T.; Van Doorn, R. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Investig. Dermatol. 2023, 143, 18–25.e1. [Google Scholar] [CrossRef]
- Facchinetti, F.; Lacroix, L.; Mezquita, L.; Scoazec, J.-Y.; Loriot, Y.; Tselikas, L.; Gazzah, A.; Rouleau, E.; Adam, J.; Michiels, S.; et al. Molecular Mechanisms of Resistance to BRAF and MEK Inhibitors in BRAFV600E Non–Small Cell Lung Cancer. Eur. J. Cancer 2020, 132, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Zanrè, V.; Bellinato, F.; Cardile, A.; Passarini, C.; Di Bella, S.; Menegazzi, M. BRAF-Mutated Melanoma Cell Lines Develop Distinct Molecular Signatures After Prolonged Exposure to AZ628 or Dabrafenib: Potential Benefits of the Antiretroviral Treatments Cabotegravir or Doravirine on BRAF-Inhibitor-Resistant Cells. Int. J. Mol. Sci. 2024, 25, 11939. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Yan, C.; Yang, Y.; Sang, L.; Hao, Q.; Yao, X.; Zhang, Y.; Yu, J.; Wang, Y.; Li, X.; et al. EGF/EGFR-YAP 1/TEAD 2 Signaling Upregulates STIM 1 in Vemurafenib Resistant Melanoma Cells. FEBS J. 2024, 291, 4969–4983. [Google Scholar] [CrossRef] [PubMed]
- Mondru, A.K.; Wilkinson, B.; Aljasir, M.A.; Alrumayh, A.; Greaves, G.; Emmett, M.; Albohairi, S.; Pritchard-Jones, R.; Cross, M.J. The ERK5 Pathway in BRAFV600E Melanoma Cells Plays a Role in Development of Acquired Resistance to Dabrafenib but Not Vemurafenib. FEBS Lett. 2024, 598, 2011–2027. [Google Scholar] [CrossRef]
- Yadav, V.; Jobe, N.; Satapathy, S.R.; Mohapatra, P.; Andersson, T. Increased MARCKS Activity in BRAF Inhibitor-Resistant Melanoma Cells Is Essential for Their Enhanced Metastatic Behavior Independent of Elevated WNT5A and IL-6 Signaling. Cancers 2022, 14, 6077. [Google Scholar] [CrossRef]
- Blateau, P.; Coyaud, E.; Laurent, E.; Béganton, B.; Ducros, V.; Chauchard, G.; Vendrell, J.A.; Solassol, J. TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma. Cancers 2020, 12, 2224. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gutierrez, M.; Anders, C.K.; Ansstas, G.; Owonikoko, T.K.; Monga, V.; Forsyth, P.A.J.; Dagogo-Jack, I.; Chandra, S.; Tsai, K.K.; et al. Trial in Progress: Phase 1a/b Study of PF-07284890 (Brain-Penetrant BRAF Inhibitor) with/without Binimetinib in Patients with BRAF V600-Mutant Solid Tumors. J. Clin. Oncol. 2021, 39 (Suppl. S15), TPS3152. [Google Scholar] [CrossRef]
- Dirven, I.; Pierre, E.; Vander Mijnsbrugge, A.-S.; Vounckx, M.; Kessels, J.I.; Neyns, B. Regorafenib Combined with BRAF/MEK Inhibitors for the Treatment of Refractory Melanoma Brain Metastases. Cancers 2024, 16, 4083. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF Inhibitor PLX8394 Selectively Disrupts BRAF Dimers and RAS-Independent BRAF-Mutant-Driven Signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Schmidt-Supprian, M.; Rad, R.; Saur, D. Tissue-Specific Tumorigenesis: Context Matters. Nat. Rev. Cancer 2017, 17, 239–253. [Google Scholar] [CrossRef] [PubMed]

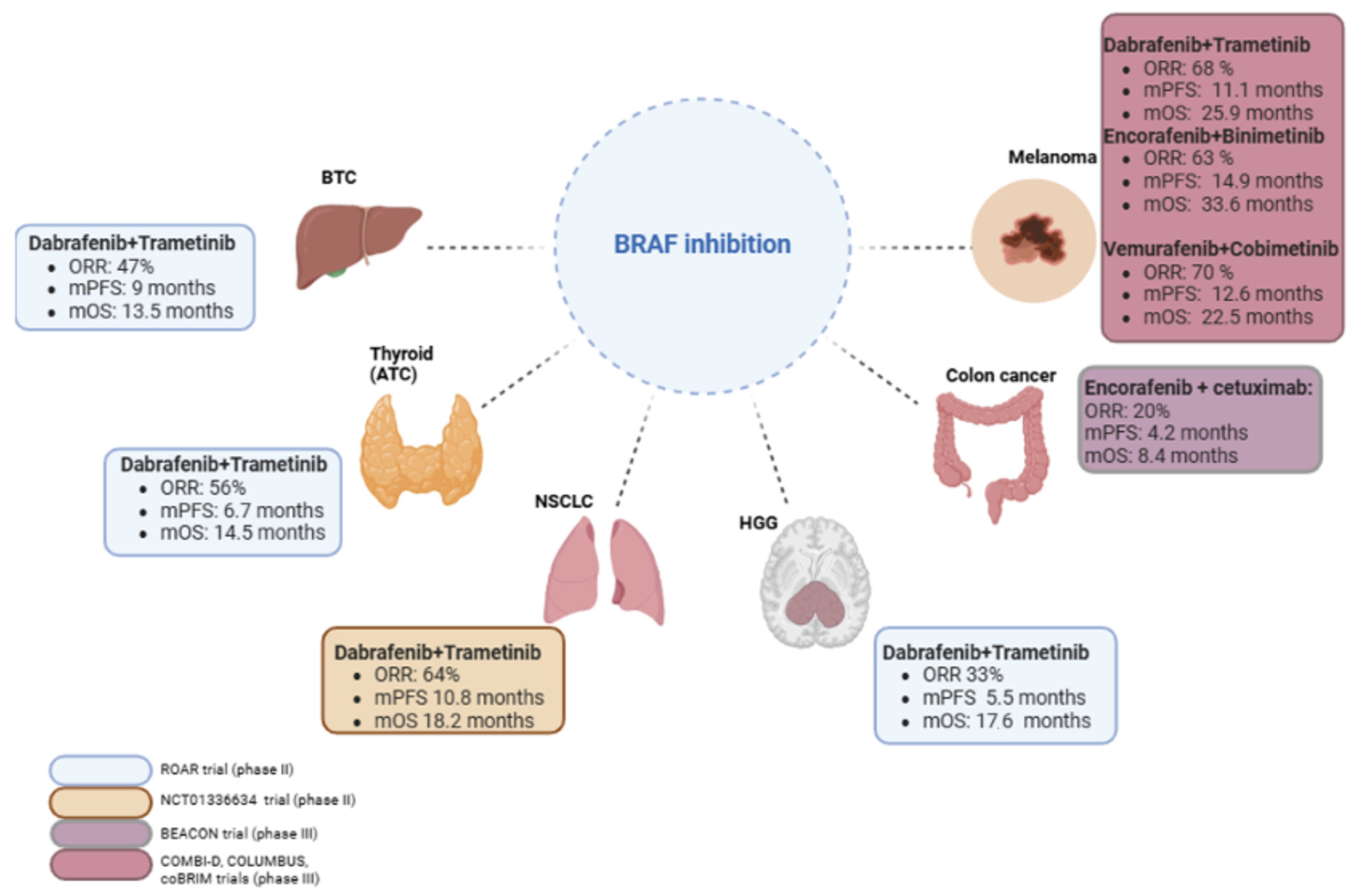

{kind=link}
{kind=link}
| Cancer Type | Treatment Regimen | Main AEs, Any Grade | Source |
|---|---|---|---|
| Metastatic melanoma | Dabrafenib + trametinib | Pyrexia (52%), alanine aminotransferase increase (10%), fatigue (27%), rash (24%), cutaneous squamous cell carcinoma (3%), decreased ejection fraction (4%) | [69] |
| Chemotherapy (dacarbazine) | Nausea (14%), vomiting (5%), fatigue (5%), diarrhea (15%), hematological toxicities (>15%) | [70] | |
| Metastatic non-small-cell lung cancer (NSCLC) | Dabrafenib + trametinib | Pyrexia (46%), nausea (40%), vomiting (35%), asthenia (28%), rash (19%), squamous cell carcinoma of skin (4%) | [18] |
| Standard second-line chemotherapy (paclitaxel, bevacizumab) | Hematological toxicity (73,4%), neuropathy (49,5%), alopecia (29,4%), vomiting (13,8%) | [71] | |
| Metastatic colorectal cancer | Encorafenib + binimetinib + cetuximab | Diarrhea (62%), acneiform dermatitis (49%), nausea (45%), vomiting (38%), pyrexia (20%) | [72] |
| Standard chemotherapy (FOLFIRI–cetuximab) | Diarrhea (48%), acneiform dermatitis (39%), nausea (41%), vomiting (29%), pyrexia (14%) | [72] |
| Tumor Type | Patient Population | Intervention | Phase | Trial Designation | Status |
|---|---|---|---|---|---|
| Melanoma | Patients with resectable stage IIIB-C BRAF V600 mutation-positive melanoma | Dabrafenib + trametinib | II | NCT01972347 | NR |
| Metastatic or unresectable melanoma carrying a BRAF V600 mutation and having relapsed on a BRAF/MEK inhibitor therapy | Dabrafenib + trametinib | I | NCT04903119 | R | |
| Advanced melanoma patients with BRAF V600E/K mutation | HL-085 + vemurafenib | II | NCT05263453 | R | |
| Unresectable BRAF-mutated stage III/IV melanoma | XL888 + vemurafenib + cobimetinib | II | NCT02721459 | NR | |
| Metastatic or unresectable melanoma carrying a BRAF V600 mutation and having relapsed on a BRAF/MEK inhibitor therapy | Nilotinib + dabrafenib/trametinib or encorafenib/binimetinib | I | NCT04903119 | R | |
| Chinese patients with stage III BRAF V600 mutation positive melanoma after complete resection | Dabrafenib + trametinib | II | NCT04666272 | R | |
| High-risk patients with stage II melanoma with BRAF mutations (COLUMBUS-AD) | Encorafenib + binimetinib | III | NCT05270044 | NR | |
| Treatment-naive patients with advanced/metastatic melanoma with BRAF alterations (STEABOARD) | Encorafenib + binimetinib + pembrolizumab | III | NCT04657991 | NR | |
| BRAF mutant metastatic melanoma (CELEBRATE) | Encorafenib + binimetinib + palbociclib | I/II | NCT04720768 | R | |
| Patients with pretreated advanced melanoma (RegoMel) | Regorafenib | II | NCT05370807 | R | |
| BRAFV600-mutated melanoma with CNS metastasis | E6201 + dabrafenib | I | NCT05388877 | NR | |
| mCRC | First-line metastatic colorectal cancer with BRAFV600E mutation | Encorafenib + cetuximab + mFOLFOX6 | III | NCT04607421 | NR |
| Metastatic colorectal cancer (mCRC) with BRAF V600E mutation after first-line treatment | HLX208 (BRAF V600E Inhibitor) + cetuximab | II | NCT04984369 | NR | |
| First-line treatment for RAS/BRAF wild-type advanced colorectal cancer | Sintilimab + cetuximab + chemotherapy | I/II | NCT06776757 | R | |
| Previously treated MSS mCRC with BRAFV600E mutation | Encorafenib + cetuximab + nivolumab | II | NCT05308446 | R | |
| Previously untreated metastatic CRC (BREAKWATER) | Encorafenib + cetuximab +/− chemotherapy | III | NCT04607421 | NR | |
| Previously untreated BRAFV600E mutant, MSI high/DMMR metastatic CRC (SEAMARK) | Encorafenib + cetuximab + pembrolizumab vs. pembrolizumab alone | II | NCT05217446 | NR | |
| BRAF V600E-mutated MSS initially resectable/potentially resectable advanced colorectal cancer | Cetuximab + encorafenib + binimetinib | II | NCT06207656 | R | |
| Thyroid | High-risk BRAFV600E-mutant differentiated thyroid carcinoma (pre-radioiodine therapy) | Vemurafenib + cobimetinib | II | NCT06440850 | R |
| Radioiodine-refractory BRAFV600E-mutant differentiated thyroid cancer (post-VEGFR TKI) | Dabrafenib + trametinib vs. cabozantinib | III | NCT06475989 | R | |
| Previously treated patients with locally advanced/metastatic, RAI-refractory BRAFV600E-mutated differentiated thyroid cancer | Dabrafenib + trametinib | III | NCT04940052 | NR | |
| Patients with metastatic radioiodine refractory BRAFV600 mutant thyroid cancer | Encorafenib + binimetinib +/− nivolumab | II | NCT04061980 | NR | |
| NSCLC | Metastatic NSCLC with BRAF V600E mutation (no prior BRAF/MEK inhibitors) | Encorafenib + binimetinib | II | NCT03915951 | NR |
| Untreated metastatic NSCLC with BRAFV600E mutation | Encorafenib + binimetinib | II | NCT04526782 | NR | |
| Patients with NSCLC, solid tumors, melanoma, high-grade gliomas | Dabrafenib + trametinib | IV | NCT03340506 | R | |
| Advanced NSCLC | Binimetinib + pembrolizumab | I | NCT03991819 | R | |
| Other solid tumors | Advanced solid tumors with non-V600E BRAF mutations (e.g., class II/III alterations) | Encorafenib + binimetinib | II | NCT03839342 | NR |
| Relapsed or progressive pediatric low-grade glioma with BRAF alterations (V600E or BRAF fusions) | Tovorafenib | II | NCT04775485 | R | |
| Advanced solid tumors with oncogenic BRAF (class I/II/III) or RAS/MAPK pathway mutations | BDTX-4933 (pan-RAF/RAS inhibitor) | I | NCT05786924 | NR | |
| adults with BRAF/NRAS-mutated advanced or metastatic solid tumors | KIN-2787 | I | NCT04913285 | R | |
| Advanced solid tumors with BRAFV600 mutations (monotherapy dose escalation; combination with trametinib in expansion) | CFT1946 (BRAF V600 degrader) ± trametinib | I/II | NCT05668585 | R | |
| Recurrent/progressive low-grade ovarian or peritoneal cavity cancer | Trametinib vs. standard of care | II/III | NCT02101788 | NR | |
| Patients with BRAF and other RAS/MAPK mutation-positive neoplasms | BDTX-4933 | I | NCT05786924 | NR | |
| Advanced solid tumors with BRAF, KRAS, and/or NRAS mutations | VS6766 +/− everolimus | I | NCT02407509 | R | |
| Advanced/metastatic malignancies harboring RAS or RAF oncogenic mutations | IMM-6-415 | I/II | NCT06208124 | R | |
| Advanced/recurrent low-grade glioma or pancreatic cancer with BRAF fusion/rearrangement | Binimetinib | II | NCT06159478 | R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mechahougui, H.; Gutmans, J.; Gouasmi, R.; Smekens, L.; Friedlaender, A. BRAF Targeting Across Solid Tumors: Molecular Aspects and Clinical Applications. Int. J. Mol. Sci. 2025, 26, 3757. https://doi.org/10.3390/ijms26083757
Mechahougui H, Gutmans J, Gouasmi R, Smekens L, Friedlaender A. BRAF Targeting Across Solid Tumors: Molecular Aspects and Clinical Applications. International Journal of Molecular Sciences. 2025; 26(8):3757. https://doi.org/10.3390/ijms26083757
Chicago/Turabian StyleMechahougui, Hiba, James Gutmans, Roumaïssa Gouasmi, Laure Smekens, and Alex Friedlaender. 2025. "BRAF Targeting Across Solid Tumors: Molecular Aspects and Clinical Applications" International Journal of Molecular Sciences 26, no. 8: 3757. https://doi.org/10.3390/ijms26083757
APA StyleMechahougui, H., Gutmans, J., Gouasmi, R., Smekens, L., & Friedlaender, A. (2025). BRAF Targeting Across Solid Tumors: Molecular Aspects and Clinical Applications. International Journal of Molecular Sciences, 26(8), 3757. https://doi.org/10.3390/ijms26083757