


Lapatinib-Resistant HER2+ Breast Cancer Cells Are Associated with Dysregulation of MAPK and p70S6K/PDCD4 Pathways and Calcium Management, Influence of Cryptotanshinone

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Results
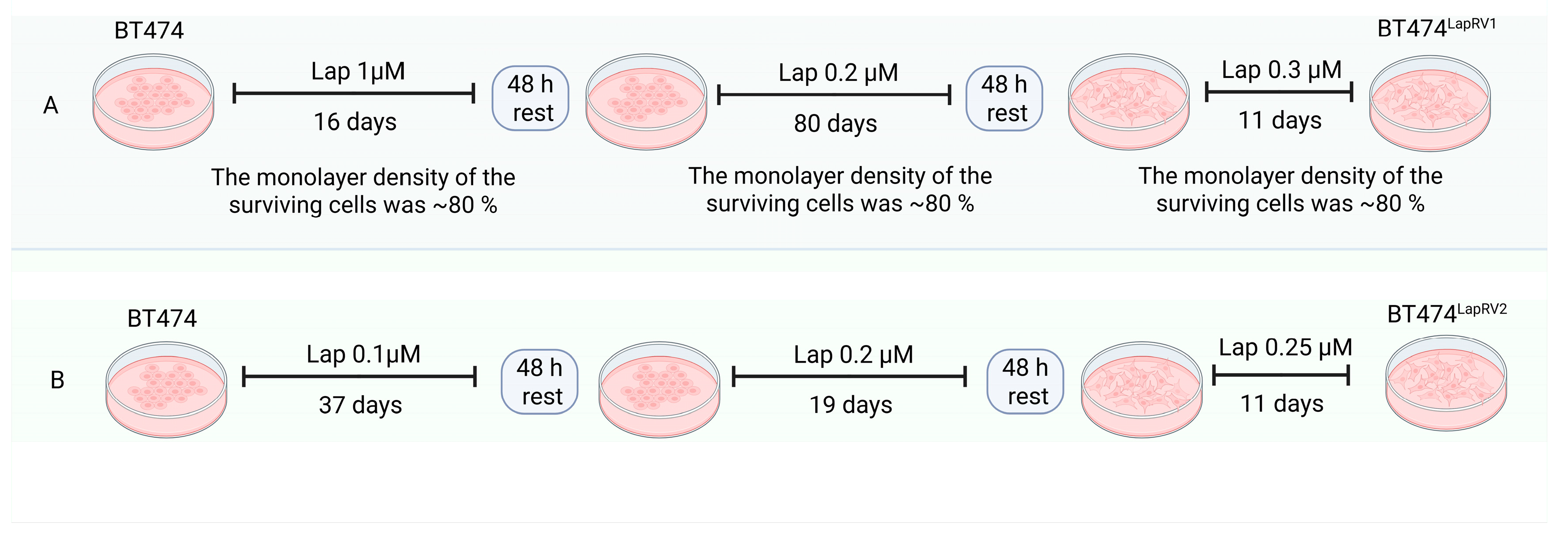
2.1. Characterization of Lap-Resistant Cell Variants
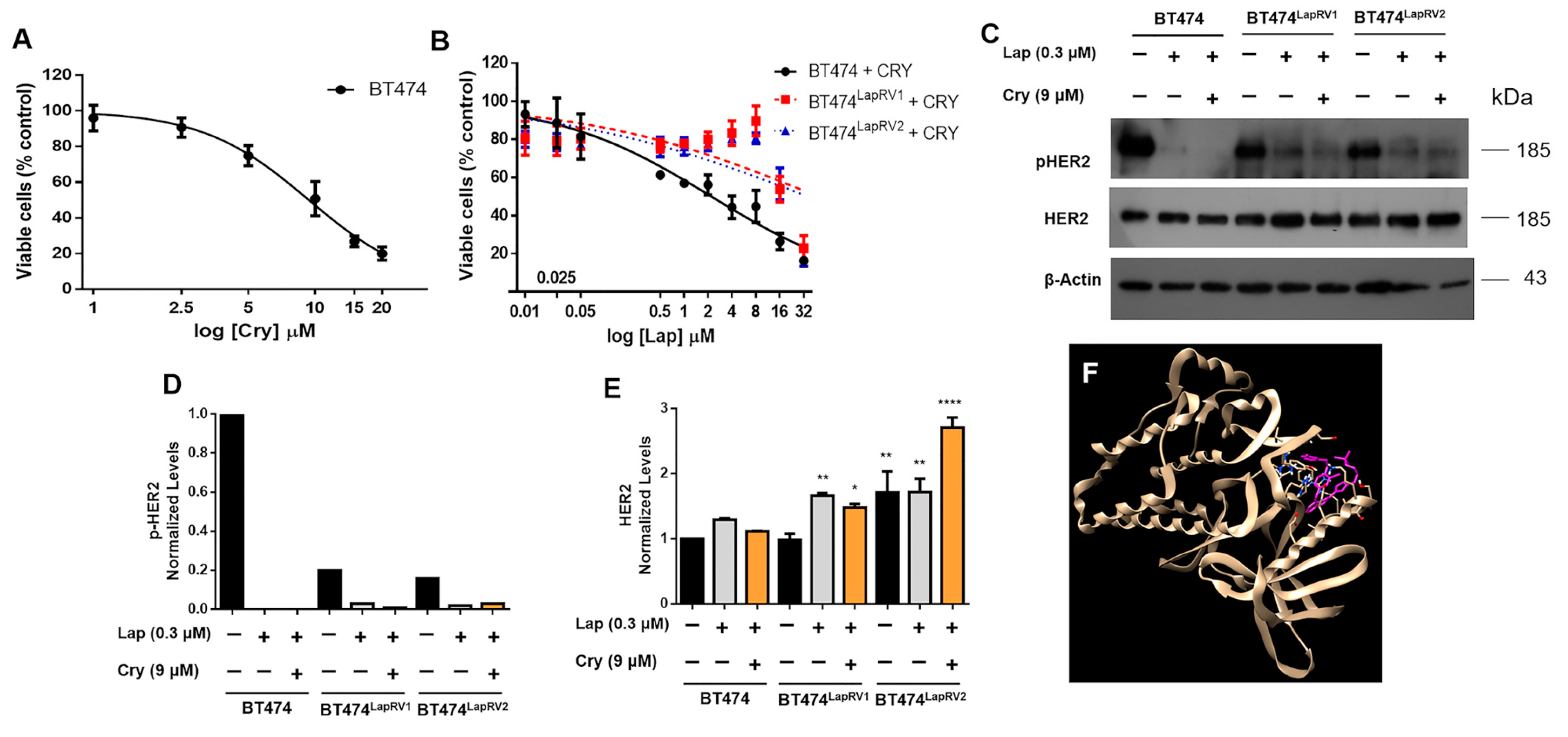
2.2. Chemoresistance in BT474 Cells Overcomes the Inhibition of HER2 Signaling
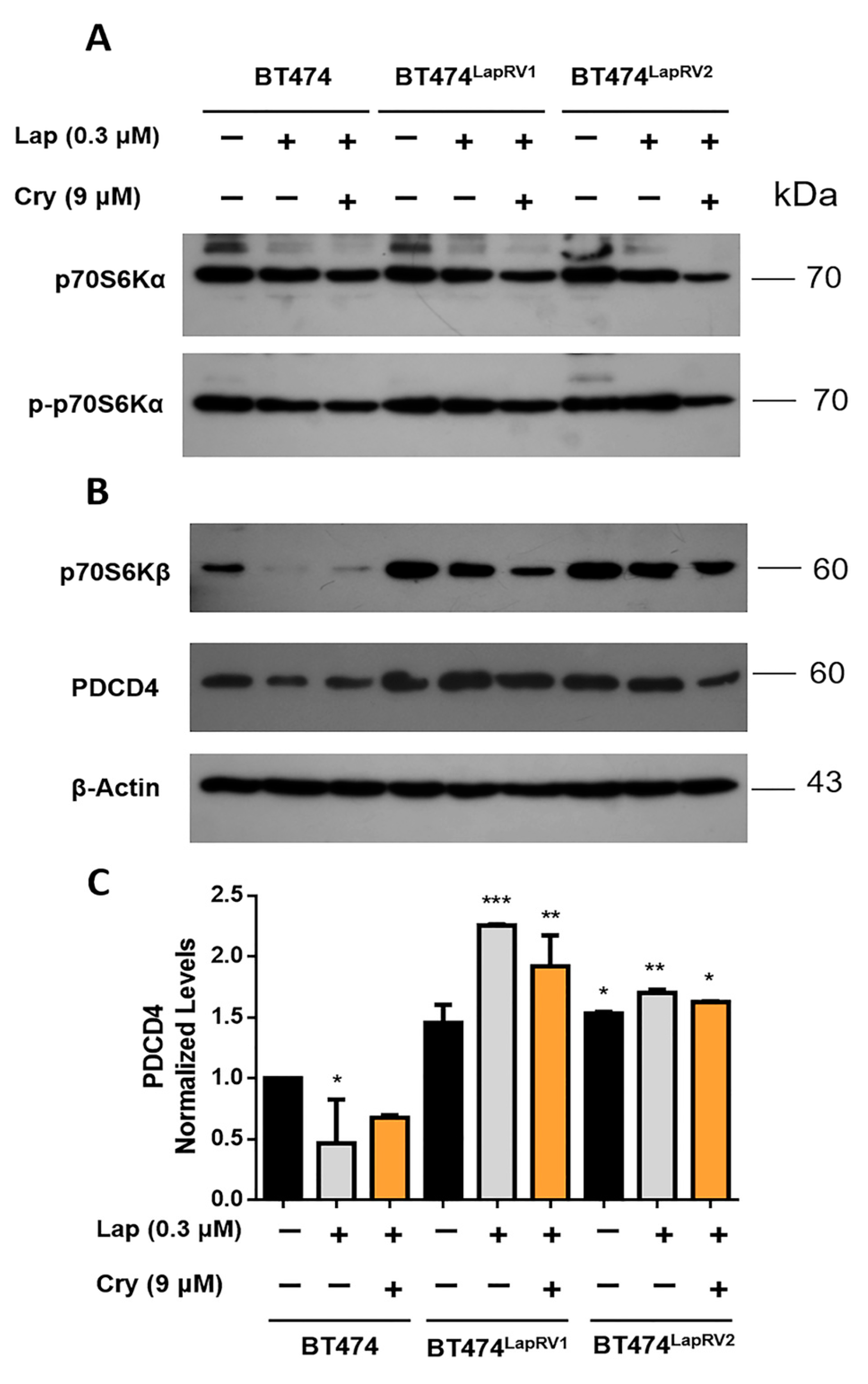
2.3. The Axis p70S6Kβ-PDCD4 Is Activated in Chemoresistant Cell Variants
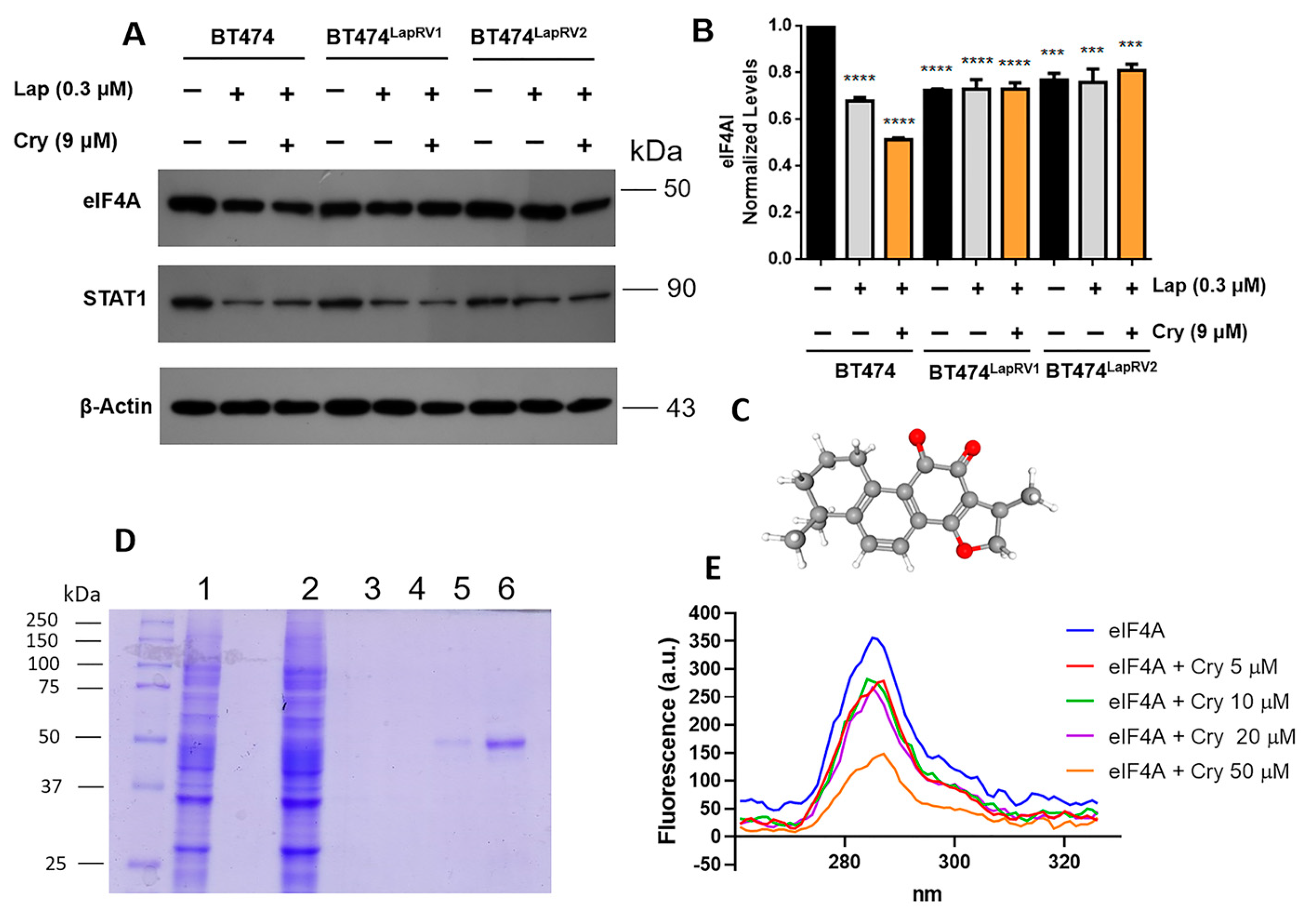
2.4. The Role of eIF4A in Chemoresistance and Its Modulation by Crytotanshinone
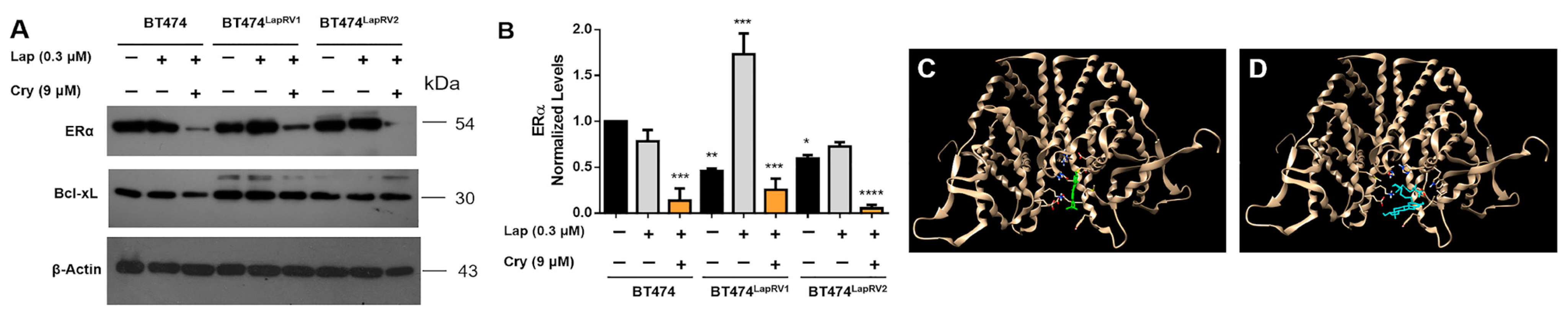
2.5. Cryptotanshinone Modulates the Estrogen Receptor (ERα)
2.6. Dysregulation of Survival Pathways in Chemoresistance Variants
2.7. Chemoresistance Is Associated with Optimized Calcium Management
2.8. NF-κB Pathway and Connection with Chemoresistance
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines and Cell Culture
4.3. Generation of the Lapatinib-Resistant BT474 Cell Line Variants (BT474LapRV1 and BT474LapRV2)
4.4. Cell Viability Assay
4.5. Colony Formation Assay
4.6. Western Blot Assays
4.7. Small Interfering RNA (siRNA)
4.8. eIF4AI Overexpression and Purification
4.9. Interaction Assay
4.10. Molecular Docking
4.11. Intracellular Calcium Quantification
4.12. Zymogram Assays
4.13. Kaplan–Meier Plotter Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, L.; Huang, Y.; Zhuo, W.; Zhu, Y.; Zhu, B.; Chen, Z. Identification and characterization of biomarkers and their functions for Lapatinib-resistant breast cancer. Med. Oncol. 2017, 34, 89. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, V.; Raimondo, L.; Formisano, L.; Giuliano, M.; De Placido, S.; Rosa, R.; Bianco, R. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat. Rev. 2015, 41, 877–883. [Google Scholar] [CrossRef]
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef] [PubMed]
- Malka-Mahieu, H.; Newman, M.; Desaubry, L.; Robert, C.; Vagner, S. Molecular Pathways: The eIF4F Translation Initiation Complex-New Opportunities for Cancer Treatment. Clin. Cancer Res. 2017, 23, 21–25. [Google Scholar] [CrossRef]
- Wang, S.; Darini, C.; Desaubry, L.; Koromilas, A.E. STAT1 Promotes KRAS Colon Tumor Growth and Susceptibility to Pharmacological Inhibition of Translation Initiation Factor eIF4A. Mol. Cancer Ther. 2016, 15, 3055–3063. [Google Scholar] [CrossRef]
- Gerson-Gurwitz, A.; Young, N.P.; Goel, V.K.; Eam, B.; Stumpf, C.R.; Chen, J.; Fish, S.; Barrera, M.; Sung, E.; Staunton, J.; et al. Zotatifin, an eIF4A-Selective Inhibitor, Blocks Tumor Growth in Receptor Tyrosine Kinase Driven Tumors. Front. Oncol. 2021, 11, 766298. [Google Scholar] [CrossRef]
- Ocana, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef]
- Szoor, A.; Toth, G.; Zsebik, B.; Szabo, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, X.; Fan, Y.; Zhou, L.; Duan, Y.; Li, S.; Sun, Z.; Zhang, C.; Yang, H.; Yuan, W.; et al. Reactivation of MAPK-SOX2 pathway confers ferroptosis sensitivity in KRAS(G12C) inhibitor resistant tumors. Redox Biol. 2024, 78, 103419. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Hou, J.; Jiang, Z.; Lin, Q. Mechanisms of resistance to CAR-T cell therapy in multiple myeloma: Latest updates from the 2024 ASH annual meeting. Exp. Hematol. Oncol. 2025, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kang, S.K.; Kwon, W.S.; Jeong, I.; Kim, T.S.; Yu, S.Y.; Cho, S.W.; Chung, H.C.; Rha, S.Y. Novel HER2-targeted therapy to overcome trastuzumab resistance in HER2-amplified gastric cancer. Sci. Rep. 2023, 13, 22648. [Google Scholar] [CrossRef]
- Elster, N.; Cremona, M.; Morgan, C.; Toomey, S.; Carr, A.; O’Grady, A.; Hennessy, B.T.; Eustace, A.J. A preclinical evaluation of the PI3K alpha/delta dominant inhibitor BAY 80-6946 in HER2-positive breast cancer models with acquired resistance to the HER2-targeted therapies trastuzumab and lapatinib. Breast Cancer Res. Treat. 2015, 149, 373–383. [Google Scholar] [CrossRef]
- Wu, Y.H.; Wu, Y.R.; Li, B.; Yan, Z.Y. Cryptotanshinone: A review of its pharmacology activities and molecular mechanisms. Fitoterapia 2020, 145, 104633. [Google Scholar] [CrossRef]
- Chang, F.W.; Fan, H.C.; Liu, J.M.; Fan, T.P.; Jing, J.; Yang, C.L.; Hsu, R.J. Estrogen Enhances the Expression of the Multidrug Transporter Gene ABCG2-Increasing Drug Resistance of Breast Cancer Cells through Estrogen Receptors. Int. J. Mol. Sci. 2017, 18, 163. [Google Scholar] [CrossRef]
- Sridharan, S.; Robeson, M.; Bastihalli-Tukaramrao, D.; Howard, C.M.; Subramaniyan, B.; Tilley, A.M.C.; Tiwari, A.K.; Raman, D. Targeting of the Eukaryotic Translation Initiation Factor 4A Against Breast Cancer Stemness. Front. Oncol. 2019, 9, 1311. [Google Scholar] [CrossRef]
- Xia, C.; Bai, X.; Hou, X.; Gou, X.; Wang, Y.; Zeng, H.; Huang, M.; Jin, J. Cryptotanshinone Reverses Cisplatin Resistance of Human Lung Carcinoma A549 Cells through Down-Regulating Nrf2 Pathway. Cell Physiol. Biochem. 2015, 37, 816–824. [Google Scholar] [CrossRef]
- Xu, Z.; Jiang, H.; Zhu, Y.; Wang, H.; Jiang, J.; Chen, L.; Xu, W.; Hu, T.; Cho, C.H. Cryptotanshinone induces ROS-dependent autophagy in multidrug-resistant colon cancer cells. Chem. Biol. Interact. 2017, 273, 48–55. [Google Scholar] [CrossRef]
- Li, H.; Gao, C.; Liang, Q.; Liu, C.; Liu, L.; Zhuang, J.; Yang, J.; Zhou, C.; Feng, F.; Sun, C. Cryptotanshinone Is a Intervention for ER-Positive Breast Cancer: An Integrated Approach to the Study of Natural Product Intervention Mechanisms. Front. Pharmacol. 2020, 11, 592109. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ortiz, A.; Pulido-Capiz, A.; Castaneda-Sanchez, C.Y.; Ibarra-Lopez, E.; Galindo-Hernandez, O.; Calderon-Fernandez, M.A.; Lopez-Cossio, L.Y.; Diaz-Molina, R.; Chimal-Vega, B.; Serafin-Higuera, N.; et al. eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model. Cells 2022, 11, 4069. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Sanchez, C.Y.; Chimal-Vega, B.; Leon-Gutierrez, R.; Araiza-Robles, A.E.; Serafin-Higuera, N.; Pulido-Capiz, A.; Rivero, I.A.; Diaz-Molina, R.; Alatorre-Meda, M.; Rodriguez-Velazquez, E.; et al. Low-Density Lipoproteins Increase Proliferation, Invasion, and Chemoresistance via an Exosome Autocrine Mechanism in MDA-MB-231 Chemoresistant Cells. Biomedicines 2024, 12, 742. [Google Scholar] [CrossRef]
- Guevara-Olaya, L.; Chimal-Vega, B.; Castaneda-Sanchez, C.Y.; Lopez-Cossio, L.Y.; Pulido-Capiz, A.; Galindo-Hernandez, O.; Diaz-Molina, R.; Ruiz Esparza-Cisneros, J.; Garcia-Gonzalez, V. LDL Promotes Disorders in β-Cell Cholesterol Metabolism, Implications on Insulin Cellular Communication Mediated by EVs. Metabolites 2022, 12, 754. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Coello, B.; Luna-Reyes, I.; Mendez-Acevedo, K.M.; Bravo-Martinez, J.; Montalvan-Sorrosa, D.; Mas-Oliva, J. Analysis of cholesterol-recognition motifs of the plasma membrane Ca2+-ATPase. J. Bioenerg. Biomembr. 2024, 56, 205–219. [Google Scholar] [CrossRef]
- Jeong, J.; VanHouten, J.N.; Dann, P.; Kim, W.; Sullivan, C.; Yu, H.; Liotta, L.; Espina, V.; Stern, D.F.; Friedman, P.A.; et al. PMCA2 regulates HER2 protein kinase localization and signaling and promotes HER2-mediated breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E282–E290. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Hurwitz, H.I.; Dees, E.C.; Dowlati, A.; Blackwell, K.L.; O’Neil, B.; Marcom, P.K.; Ellis, M.J.; Overmoyer, B.; Jones, S.F.; et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J. Clin. Oncol. 2005, 23, 5305–5313. [Google Scholar] [CrossRef]
- Bakhsh, T.; Alyami, N.M. Inducing breast cancer cell death: The impact of taxodone on proliferation through apoptosis. Heliyon 2024, 10, e34044. [Google Scholar] [CrossRef]
- Cerezo, M.; Guemiri, R.; Druillennec, S.; Girault, I.; Malka-Mahieu, H.; Shen, S.; Allard, D.; Martineau, S.; Welsch, C.; Agoussi, S.; et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat. Med. 2018, 24, 1877–1886. [Google Scholar] [CrossRef]
- Giuliano, M.; Hu, H.; Wang, Y.C.; Fu, X.; Nardone, A.; Herrera, S.; Mao, S.; Contreras, A.; Gutierrez, C.; Wang, T.; et al. Upregulation of ER Signaling as an Adaptive Mechanism of Cell Survival in HER2-Positive Breast Tumors Treated with Anti-HER2 Therapy. Clin. Cancer Res. 2015, 21, 3995–4003. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Lochhead, P.A. ERK5 Signalling and Resistance to ERK1/2 Pathway Therapeutics: The Path Less Travelled? Front. Cell Dev. Biol. 2022, 10, 839997. [Google Scholar] [CrossRef] [PubMed]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol. Cell. Biol. 2012, 32, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, J.; Yan, X.; Liu, Q.; Zhang, M. TROP2 promotes the proliferation of triple-negative breast cancer cells via calcium ion-dependent ER stress signaling pathway. Cell Biochem. Biophys. 2024, 82, 2205–2216. [Google Scholar] [CrossRef]
- Acosta-Montano, P.; Rodriguez-Velazquez, E.; Ibarra-Lopez, E.; Frayde-Gomez, H.; Mas-Oliva, J.; Delgado-Coello, B.; Rivero, I.A.; Alatorre-Meda, M.; Aguilera, J.; Guevara-Olaya, L.; et al. Fatty Acid and Lipopolysaccharide Effect on Beta Cells Proteostasis and Its Impact on Insulin Secretion. Cells 2019, 8, 884. [Google Scholar] [CrossRef]
- Curry, M.C.; Luk, N.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Distinct regulation of cytoplasmic calcium signals and cell death pathways by different plasma membrane calcium ATPase isoforms in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2012, 287, 28598–28608. [Google Scholar] [CrossRef]
- Song, K.Y.; Zhang, X.Z.; Li, F.; Ji, Q.R. Silencing of ATP2B1-AS1 contributes to protection against myocardial infarction in mouse via blocking NFKBIA-mediated NF-κB signalling pathway. J. Cell. Mol. Med. 2020, 24, 4466–4479. [Google Scholar] [CrossRef]
- Tian, M.; Tian, D.; Qiao, X.; Li, J.; Zhang, L. Modulation of Myb-induced NF-kB-STAT3 signaling and resulting cisplatin resistance in ovarian cancer by dietary factors. J. Cell Physiol. 2019, 234, 21126–21134. [Google Scholar] [CrossRef]
- Xia, J.; Matsuhashi, S.; Hamajima, H.; Iwane, S.; Takahashi, H.; Eguchi, Y.; Mizuta, T.; Fujimoto, K.; Kuroda, S.; Ozaki, I. The role of PKC isoforms in the inhibition of NF-κB activation by vitamin K2 in human hepatocellular carcinoma cells. J. Nutr. Biochem. 2012, 23, 1668–1675. [Google Scholar] [CrossRef]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef]
- Zhang, R.; Qiao, H.; Chen, S.; Chen, X.; Dou, K.; Wei, L.; Zhang, J. Berberine reverses lapatinib resistance of HER2-positive breast cancer cells by increasing the level of ROS. Cancer Biol. Ther. 2016, 17, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Zhang, J.; Tsai, M.H.; Yu, D. PI3K-independent mTOR activation promotes lapatinib resistance and IAP expression that can be effectively reversed by mTOR and Hsp90 inhibition. Cancer Biol. Ther. 2015, 16, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Colomer, R.; Brunet, J.; Menendez, J.A. Low-scale phosphoproteome analyses identify the mTOR effector p70 S6 kinase 1 as a specific biomarker of the dual-HER1/HER2 tyrosine kinase inhibitor lapatinib (Tykerb) in human breast carcinoma cells. Ann. Oncol. 2008, 19, 1097–1109. [Google Scholar] [CrossRef]
- Fenton, T.R.; Gout, I.T. Functions and regulation of the 70 kDa ribosomal S6 kinases. Int. J. Biochem. Cell Biol. 2011, 43, 47–59. [Google Scholar] [CrossRef]
- Cai, Q.; Yang, H.S.; Li, Y.C.; Zhu, J. Dissecting the Roles of PDCD4 in Breast Cancer. Front. Oncol. 2022, 12, 855807. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Herrera-Montavez, C.; Montserrat-Sangra, S.; Araiza-Olivera, D.; Hu, J.; Neumann-Domer, R.; Kuruvilla, M.; Bellacosa, A.; Testa, J.R.; Jin, J.; et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal 2022, 15, eabn2707. [Google Scholar] [CrossRef]
- Chen, W.; Pan, Y.; Wang, S.; Liu, Y.; Chen, G.; Zhou, L.; Ni, W.; Wang, A.; Lu, Y. Cryptotanshinone activates AMPK-TSC2 axis leading to inhibition of mTORC1 signaling in cancer cells. BMC Cancer 2017, 17, 34. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- VanHouten, J.; Sullivan, C.; Bazinet, C.; Ryoo, T.; Camp, R.; Rimm, D.L.; Chung, G.; Wysolmerski, J. PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 11405–11410. [Google Scholar] [CrossRef]
- Park, K.C.; Kim, J.M.; Kim, S.Y.; Kim, S.M.; Lim, J.H.; Kim, M.K.; Fang, S.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. PMCA inhibition reverses drug resistance in clinically refractory cancer patient-derived models. BMC Med. 2023, 21, 38. [Google Scholar] [CrossRef]
- Souto-Guevara, C.A.; Obiol, D.; Bruno, C.L.; Ferreira-Gomes, M.S.; Rossi, J.; Costabel, M.D.; Mangialavori, I.C. Magnesium enhances aurintricarboxylic acid’s inhibitory action on the plasma membrane Ca2+-ATPase. Sci. Rep. 2024, 14, 14693. [Google Scholar] [CrossRef] [PubMed]
- Presti, D.; Quaquarini, E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2? Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. [Google Scholar] [CrossRef] [PubMed]
- Pulido-Capiz, A.; Chimal-Vega, B.; Avila-Barrientos, L.P.; Campos-Valenzuela, A.; Diaz-Molina, R.; Muniz-Salazar, R.; Galindo-Hernandez, O.; Garcia-Gonzalez, V. Auraptene Boosts the Efficacy of the Tamoxifen Metabolites Endoxifen and 4-OH-Tamoxifen in a Chemoresistant ER+ Breast Cancer Model. Pharmaceutics 2024, 16, 1179. [Google Scholar] [CrossRef]
- Mezynski, M.J.; Farrelly, A.M.; Cremona, M.; Carr, A.; Morgan, C.; Workman, J.; Armstrong, P.; McAuley, J.; Madden, S.; Fay, J.; et al. Targeting the PI3K and MAPK pathways to improve response to HER2-targeted therapies in HER2-positive gastric cancer. J. Transl. Med. 2021, 19, 184. [Google Scholar] [CrossRef] [PubMed]
- Wulandari, F.; Fauzi, A.; Da’i, M.; Mirzaei, M.; Maryati; Harismah, K. Screening and identification of potential target of 1′-acetoxychavicol acetate (ACA) in acquired lapatinib-resistant breast cancer. Heliyon 2024, 10, e40769. [Google Scholar] [CrossRef]
- Zheng, S.; Chen, R.; Zhang, L.; Tan, L.; Li, L.; Long, F.; Wang, T. Unraveling the future: Innovative design strategies and emerging challenges in HER2-targeted tyrosine kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2024, 276, 116702. [Google Scholar] [CrossRef]
- Medina, P.J.; Goodin, S. Lapatinib: A dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin. Ther. 2008, 30, 1426–1447. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Patel, H.; Porter, R.H.; Palmer, A.M.; Croucher, M.J. Comparison of human recombinant adenosine A2B receptor function assessed by Fluo-3-AM fluorometry and microphysiometry. Br. J. Pharmacol. 2003, 138, 671–677. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Variant | BT474 | BT474LapRV1 | BT474LapRV2 |
|---|---|---|---|
| IC50 Lap | 3.16 ± 0.06 | 34.72 ± 0.11 | 23.3 ± 0.13 |
| IC50 Lap + Cry | 2.2 ± 0.04 | 48.28 ± 0.09 | 36.04 ± 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Valencia, J.; García-Villarreal, R.; Rodríguez-Jiménez, M.; Hernández-Avalos, A.D.; Rivero, I.A.; Vique-Sánchez, J.L.; Chimal-Vega, B.; Pulido-Capiz, A.; García-González, V. Lapatinib-Resistant HER2+ Breast Cancer Cells Are Associated with Dysregulation of MAPK and p70S6K/PDCD4 Pathways and Calcium Management, Influence of Cryptotanshinone. Int. J. Mol. Sci. 2025, 26, 3763. https://doi.org/10.3390/ijms26083763
Hernández-Valencia J, García-Villarreal R, Rodríguez-Jiménez M, Hernández-Avalos AD, Rivero IA, Vique-Sánchez JL, Chimal-Vega B, Pulido-Capiz A, García-González V. Lapatinib-Resistant HER2+ Breast Cancer Cells Are Associated with Dysregulation of MAPK and p70S6K/PDCD4 Pathways and Calcium Management, Influence of Cryptotanshinone. International Journal of Molecular Sciences. 2025; 26(8):3763. https://doi.org/10.3390/ijms26083763
Chicago/Turabian StyleHernández-Valencia, Jorge, Ruth García-Villarreal, Manuel Rodríguez-Jiménez, Alex Daniel Hernández-Avalos, Ignacio A. Rivero, José Luis Vique-Sánchez, Brenda Chimal-Vega, Angel Pulido-Capiz, and Victor García-González. 2025. "Lapatinib-Resistant HER2+ Breast Cancer Cells Are Associated with Dysregulation of MAPK and p70S6K/PDCD4 Pathways and Calcium Management, Influence of Cryptotanshinone" International Journal of Molecular Sciences 26, no. 8: 3763. https://doi.org/10.3390/ijms26083763
APA StyleHernández-Valencia, J., García-Villarreal, R., Rodríguez-Jiménez, M., Hernández-Avalos, A. D., Rivero, I. A., Vique-Sánchez, J. L., Chimal-Vega, B., Pulido-Capiz, A., & García-González, V. (2025). Lapatinib-Resistant HER2+ Breast Cancer Cells Are Associated with Dysregulation of MAPK and p70S6K/PDCD4 Pathways and Calcium Management, Influence of Cryptotanshinone. International Journal of Molecular Sciences, 26(8), 3763. https://doi.org/10.3390/ijms26083763