
Structural and Energetic Insights into SARS-CoV-2 Evolution: Analysis of hACE2–RBD Binding in Wild-Type, Delta, and Omicron Subvariants


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Model Assessment and Validation
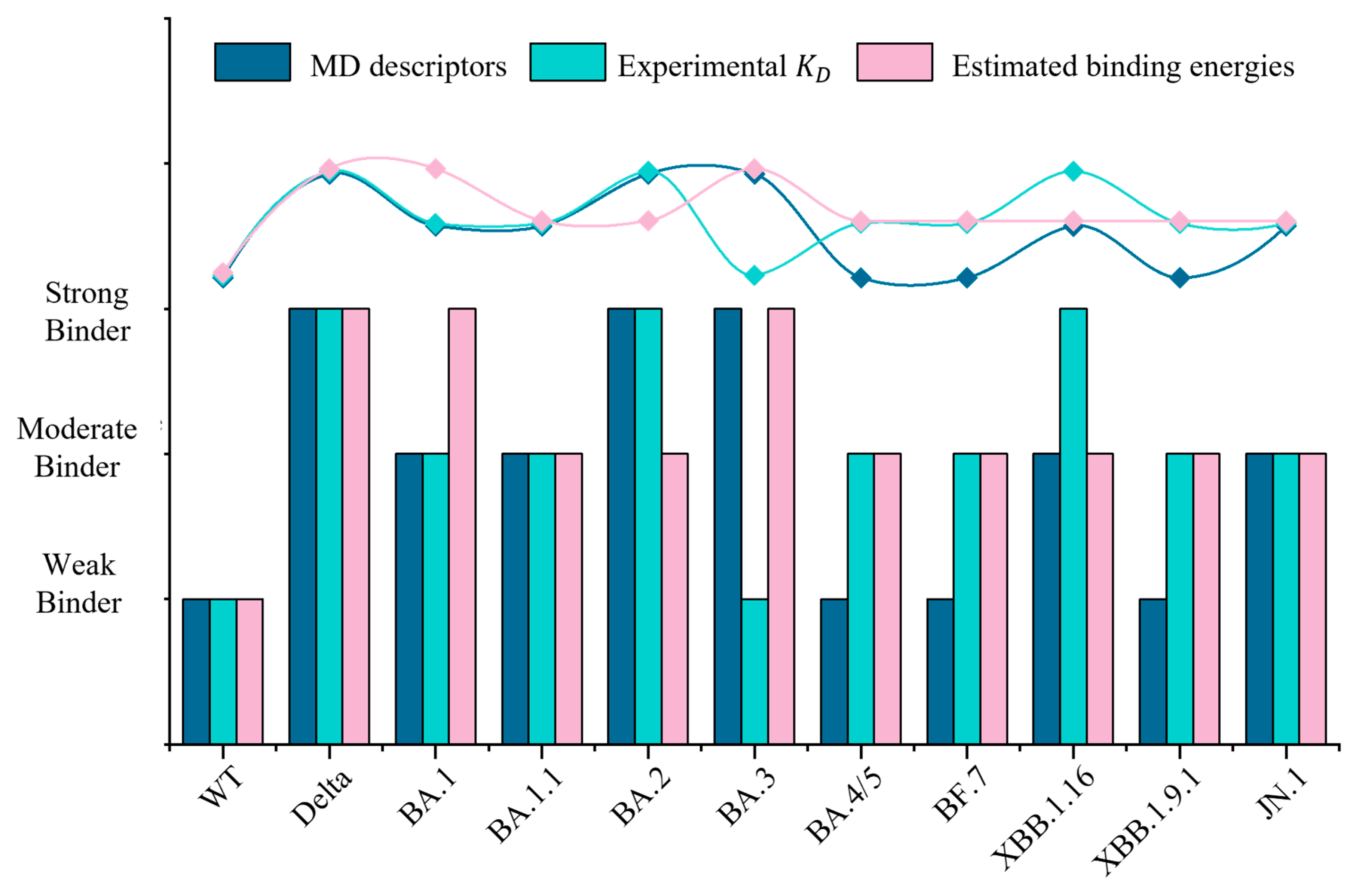
2.2. Binding Energies Reveal No Consistent Increase in Affinity to hACE2 with Evolution
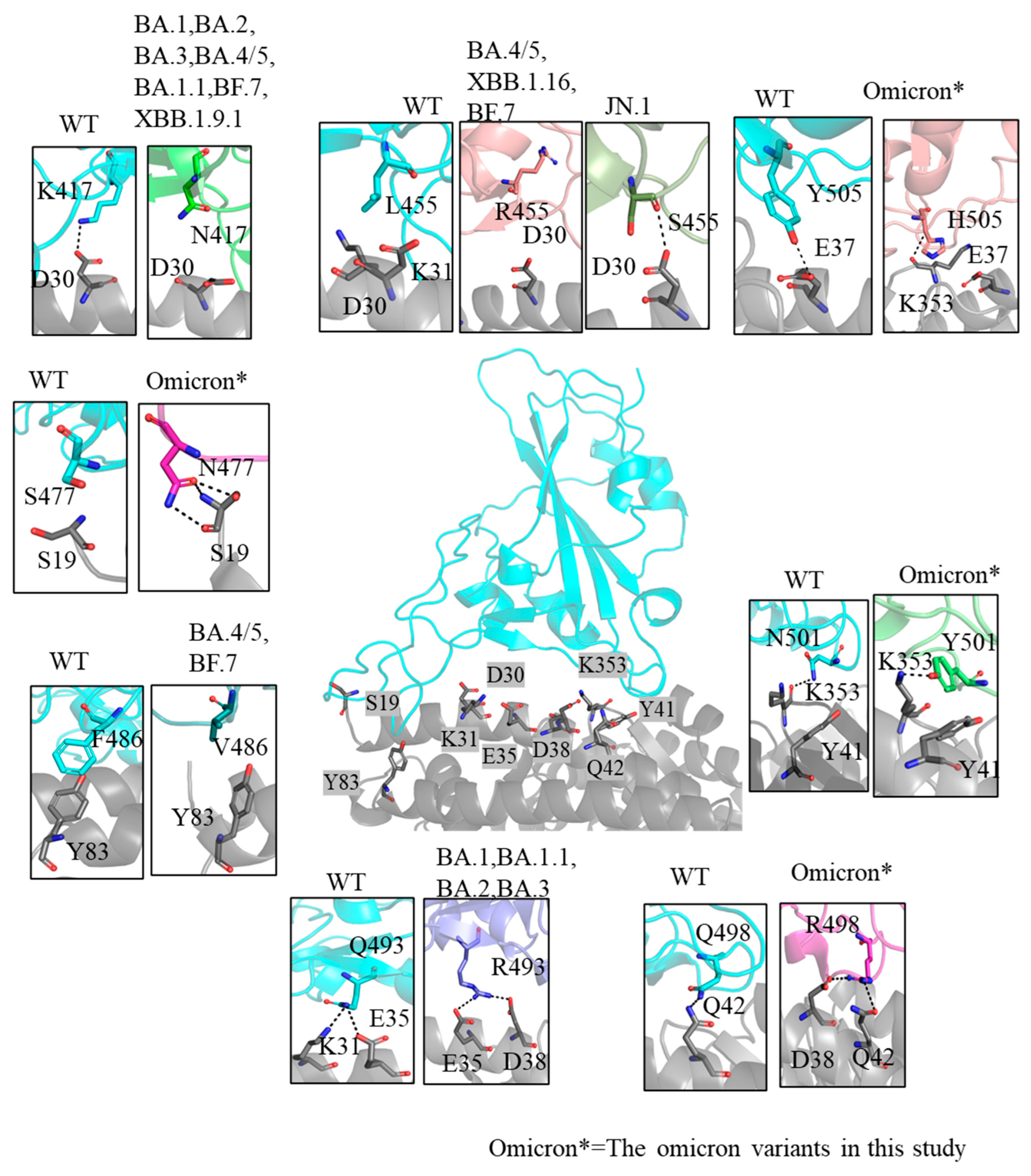
2.3. Inconsistently Enhanced Electrostatic Potential and Binding Interactions in Omicron Variants
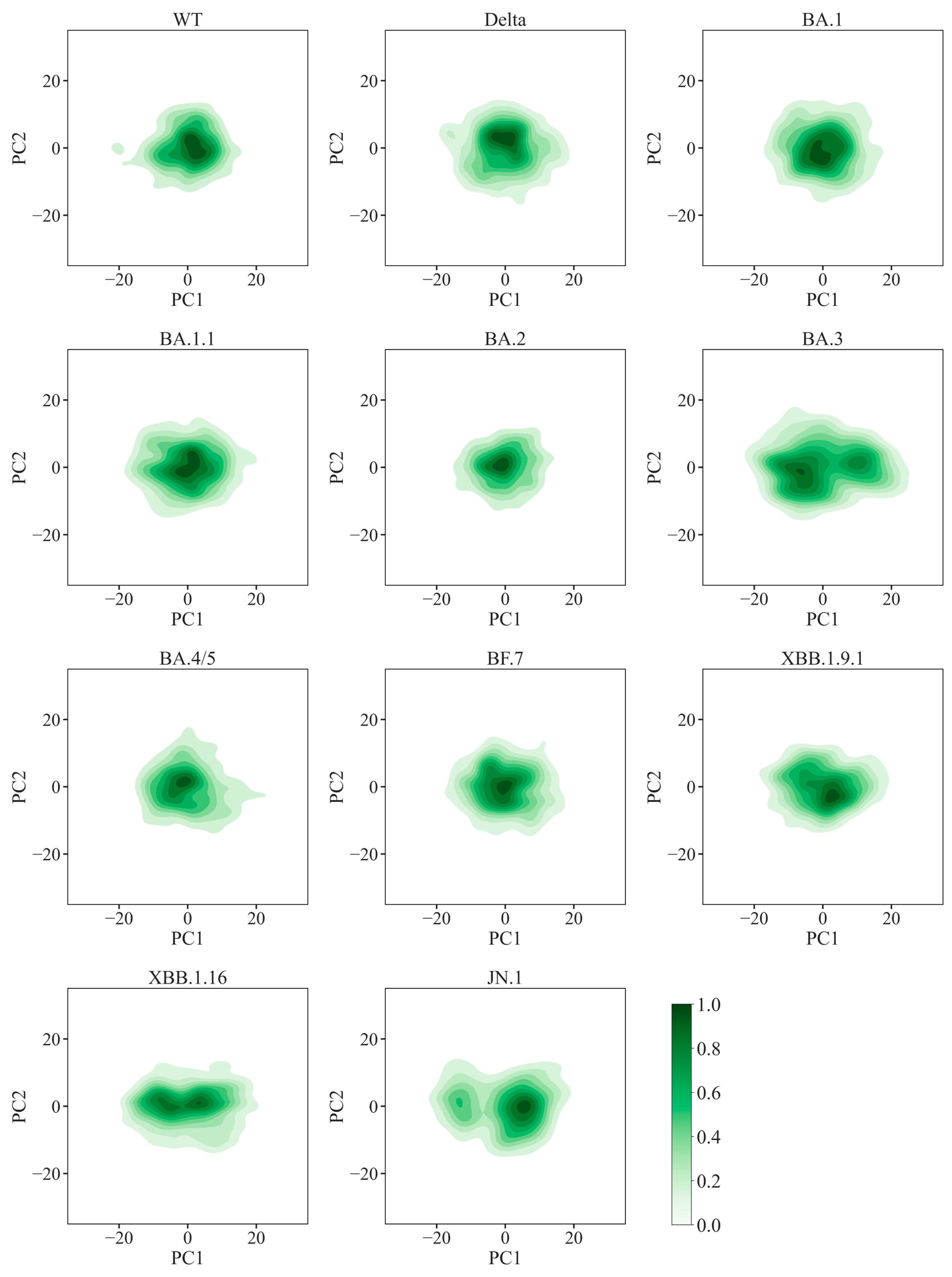
2.4. Enhanced Global Stability and Diverse Local Flexibility in Omicron Variants
2.5. Insights into Immune Evasion Features
3. Materials and Methods
3.1. hACE2–RBD Complex Structure Modeling
3.2. Molecular Dynamics Simulations of hACE2–RBD Complexes
3.3. MM/GBSA Interaction Energy Calculations
3.4. PMF-Based Binding Free Energy Calculations
3.5. Principal Component and Free Energy Landscape Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Gili, R.; Burioni, R. SARS-CoV-2 before and after Omicron: Two different viruses and two different diseases? J. Transl. Med. 2023, 21, 251. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Weekly Epidemiological Update, Edition 86, 5 April 2022. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 20 December 2024).
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Bian, J.; Li, Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm. Sin. B 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Li, J.; Hou, C.; Wang, M.; Liao, C.; Guo, S.; Shi, L.; Ma, X.; Zhang, H.; Jiang, S.; Zheng, B. Hydrophobic interaction determines docking affinity of SARS-CoV-2 variants with antibodies. arXiv 2021, arXiv:2103.00399. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Binding interactions between receptor-binding domain of spike protein and human angiotensin converting enzyme-2 in omicron variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; De Oliveira, T.; Vermeulen, M.; Van der Berg, K. SARS-CoV-2 501Y. V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Li, B.; Deng, A.; Li, K.; Hu, Y.; Li, Z.; Shi, Y.; Xiong, Q.; Liu, Z.; Guo, Q.; Zou, L. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Nat. Commun. 2022, 13, 460. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef]
- Xue, S.; Han, Y.; Wu, F.; Wang, Q. Mutations in the SARS-CoV-2 spike receptor binding domain and their delicate balance between ACE2 affinity and antibody evasion. Protein Cell 2024, 15, 403–418. [Google Scholar] [CrossRef]
- Barozi, V.; Edkins, A.L.; Bishop, Ö.T. Evolutionary progression of collective mutations in Omicron sub-lineages towards efficient RBD-hACE2: Allosteric communications between and within viral and human proteins. Comput. Struct. Biotechnol. J. 2022, 20, 4562–4578. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, W.; Dong, W.; Xu, J. Origin and evolutionary analysis of the SARS-CoV-2 Omicron variant. J. Biosaf. Biosecur. 2022, 4, 33–37. [Google Scholar] [CrossRef]
- Berkhout, B.; Herrera-Carrillo, E. SARS-CoV-2 evolution: On the sudden appearance of the omicron variant. J. Virol. 2022, 96, e00090-22. [Google Scholar] [CrossRef]
- Wei, C.; Shan, K.-J.; Wang, W.; Zhang, S.; Huan, Q.; Qian, W. Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J. Genet. Genom. 2021, 48, 1111–1121. [Google Scholar] [CrossRef]
- Callaway, E. Heavily mutated Omicron variant puts scientists on alert. Nature 2021, 600, 21. [Google Scholar] [CrossRef]
- Parsons, R.J.; Acharya, P. Evolution of the SARS-CoV-2 Omicron spike. Cell Rep. 2023, 42, 113444. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hannon, W.W.; Loes, A.N.; Hauser, K.; Dillen, J.R.; Ferri, E.; Farrell, A.G.; Dadonaite, B.; McCallum, M. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science 2022, 377, 420–424. [Google Scholar] [CrossRef]
- Gao, X.; Wang, F.; Liu, H.; Chai, J.; Tian, G.; Yao, L.; Chen, C.; Huo, P.; Yao, Y.; Wen, J. BF. 7: A new Omicron subvariant characterized by rapid transmission. Clin. Microbiol. Infect. 2024, 30, 137–141. [Google Scholar] [CrossRef]
- Lu, Y.; Ao, D.; He, X.; Wei, X. The rising SARS-CoV-2 JN. 1 variant: Evolution, infectivity, immune escape, and response strategies. MedComm 2024, 5, e675. [Google Scholar] [CrossRef]
- Looi, M.-K. What do we know about the Arcturus XBB. 1.16 subvariant? BMJ 2023, 381, 1074. [Google Scholar] [CrossRef] [PubMed]
- Abeywardhana, S.; Premathilaka, M.; Bandaranayake, U.; Perera, D.; Peiris, L.D.C. In silico study of SARS-CoV-2 spike protein RBD and human ACE-2 affinity dynamics across variants and Omicron subvariants. J. Med. Virol. 2023, 95, e28406. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef]
- Celik, I.; Yadav, R.; Duzgun, Z.; Albogami, S.; El-Shehawi, A.M.; Idroes, R.; Tallei, T.E.; Emran, T.B. Interactions of the receptor binding domain of SARS-CoV-2 variants with hACE2: Insights from molecular docking analysis and molecular dynamic simulation. Biology 2021, 10, 880. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D. Structural diversity of the SARS-CoV-2 Omicron spike. Mol. Cell 2022, 82, 2050–2068. [Google Scholar] [CrossRef]
- Yang, H.; Guo, H.; Wang, A.; Cao, L.; Fan, Q.; Jiang, J.; Wang, M.; Lin, L.; Ge, X.; Wang, H. Structural basis for the evolution and antibody evasion of SARS-CoV-2 BA. 2.86 and JN. 1 subvariants. Nat. Commun. 2024, 15, 7715. [Google Scholar] [CrossRef]
- Ramakrishnan, C.; Ramachandran, G. Stereochemical criteria for polypeptide and protein chain conformations: II. Allowed conformations for a pair of peptide units. Biophys. J. 1965, 5, 909–933. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct. Target. Ther. 2022, 7, 8. [Google Scholar] [CrossRef]
- Shah, M.; Woo, H.G. Omicron: A heavily mutated SARS-CoV-2 variant exhibits stronger binding to ACE2 and potently escapes approved COVID-19 therapeutic antibodies. Front. Immunol. 2022, 12, 830527. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; He, X.; Ren, Y.; Wang, Z.; Zhou, H.; Fan, S.; Zhu, C.; Liu, D.; Shao, B.; Liu, T.-Y. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022, 32, 593–595. [Google Scholar] [CrossRef]
- You, W.; Tang, Z.; Chang, C.-E.A. Potential mean force from umbrella sampling simulations: What can we learn and what is missed? J. Chem. Theory Comput. 2019, 15, 2433–2443. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Zhang, J.Z.; He, X. Calculation of protein–ligand binding affinities based on a fragment quantum mechanical method. RSC Adv. 2015, 5, 107020–107030. [Google Scholar] [CrossRef]
- Sang, P.; Chen, Y.-Q.; Liu, M.-T.; Wang, Y.-T.; Yue, T.; Li, Y.; Yin, Y.-R.; Yang, L.-Q. Electrostatic interactions are the primary determinant of the binding affinity of SARS-CoV-2 spike RBD to ACE2: A computational case study of omicron variants. Int. J. Mol. Sci. 2022, 23, 14796. [Google Scholar] [CrossRef] [PubMed]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.-D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- Kodchakorn, K.; Kongtawelert, P. Molecular dynamics study on the strengthening behavior of Delta and Omicron SARS-CoV-2 spike RBD improved receptor-binding affinity. PLoS ONE 2022, 17, e0277745. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Jafary, F.; Joozdani, F.A.; Shahzamani, K.; Jafari, S.; Mirhendi, H.; Ganjalikhany, M.R. Different aspects in explaining how mutations could affect the binding mechanism of receptor binding domain of SARS-CoV-2 spike protein in interaction with ACE2. PLoS ONE 2023, 18, e0291210. [Google Scholar] [CrossRef]
- Morozov, A.V.; Kortemme, T. Potential functions for hydrogen bonds in protein structure prediction and design. Adv. Protein Chem. 2005, 72, 1–38. [Google Scholar] [CrossRef]
- Philip, A.M.; Ahmed, W.S.; Biswas, K.H. Reversal of the unique Q493R mutation increases the affinity of Omicron S1-RBD for ACE2. Comput. Struct. Biotechnol. J. 2023, 21, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X. BA. 2.12. 1, BA. 4 and BA. 5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Zhang, Q.E.; Lindenberger, J.; Parsons, R.J.; Thakur, B.; Parks, R.; Park, C.S.; Huang, X.; Sammour, S.; Janowska, K.; Spence, T.N. SARS-CoV-2 Omicron XBB lineage spike structures, conformations, antigenicity, and receptor recognition. Mol. Cell 2024, 84, 2747–2764.e7. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, Z.; Niu, T.; Xie, Y.; Zhao, Z.; Li, D.; He, Q.; Sun, W.; Shi, K.; Guo, W. Key mechanistic features of the trade-off between antibody escape and host cell binding in the SARS-CoV-2 Omicron variant spike proteins. EMBO J. 2024, 43, 1484–1498. [Google Scholar] [CrossRef]
- Jian, F.; Wang, J.; Yisimayi, A.; Song, W.; Xu, Y.; Chen, X.; Niu, X.; Yang, S.; Yu, Y.; Wang, P. Evolving antibody response to SARS-CoV-2 antigenic shift from XBB to JN. 1. Nature 2025, 637, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative binding free energy calculations in drug discovery: Recent advances and practical considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Renaud, J.-P.; Chung, C.-W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Steinkellner, G.; Köchl, K.; Gruber, K.; Gruber, C.C. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep. 2021, 11, 4320. [Google Scholar] [CrossRef]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P.; Verkhivker, G. AlphaFold2-Enabled Atomistic Modeling of Structure, Conformational Ensembles, and Binding Energetics of the SARS-CoV-2 Omicron BA. 2.86 Spike Protein with ACE2 Host Receptor and Antibodies: Compensatory Functional Effects of Binding Hotspots in Modulating Mechanisms of Receptor Binding and Immune Escape. J. Chem. Inf. Model. 2024, 64, 1657–1681. [Google Scholar] [CrossRef]
- Verkhivker, G.; Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P. Probing conformational landscapes of binding and allostery in the SARS-CoV-2 omicron variant complexes using microsecond atomistic simulations and perturbation-based profiling approaches: Hidden role of omicron mutations as modulators of allosteric signaling and epistatic relationships. Phys. Chem. Chem. Phys. 2023, 25, 21245–21266. [Google Scholar] [CrossRef]
- Li, M.; Liu, X.; Zhang, S.; Liang, S.; Zhang, Q.; Chen, J. Deciphering the binding mechanism of inhibitors of the SARS-CoV-2 main protease through multiple replica accelerated molecular dynamics simulations and free energy landscapes. Phys. Chem. Chem. Phys. 2022, 24, 22129–22143. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, D.; Lee, C.-C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; Van Gils, M.J.; Sanders, R.W. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.P.; Qu, P.; Zeng, C.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Lozanski, G.; Mallampalli, R.K.; Saif, L.J.; Oltz, E.M. Neutralization of the SARS-CoV-2 deltacron and BA.3 variants. N. Engl. J. Med. 2022, 386, 2340–2342. [Google Scholar] [CrossRef]
- Tai, W.; Yang, K.; Liu, Y.; Li, R.; Feng, S.; Chai, B.; Zhuang, X.; Qi, S.; Shi, H.; Liu, Z. A lung-selective delivery of mRNA encoding broadly neutralizing antibody against SARS-CoV-2 infection. Nat. Commun. 2023, 14, 8042. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Poloni, C.; Zhu, X.; Bezeruk, A.; Tidey, K.; Ahmed, S.; Tuttle, K.S.; Vahdatihassani, F.; Cholak, S. Altered receptor binding, antibody evasion and retention of T cell recognition by the SARS-CoV-2 XBB. 1.5 spike protein. Nat. Commun. 2024, 15, 1854. [Google Scholar] [CrossRef]
- Sun, C.; Kang, Y.-F.; Liu, Y.-T.; Kong, X.-W.; Xu, H.-Q.; Xiong, D.; Xie, C.; Liu, Y.-H.; Peng, S.; Feng, G.-K. Parallel profiling of antigenicity alteration and immune escape of SARS-CoV-2 Omicron and other variants. Signal Transduct. Target. Ther. 2022, 7, 42. [Google Scholar] [CrossRef]
- Geng, Q.; Wan, Y.; Hsueh, F.-C.; Shang, J.; Ye, G.; Bu, F.; Herbst, M.; Wilkens, R.; Liu, B.; Li, F. Lys417 acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike protein. Elife 2023, 12, e74060. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.-J.; Frenz, B.; Rottier, P.J.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Hensley, S.E.; Das, S.R.; Bailey, A.L.; Schmidt, L.M.; Hickman, H.D.; Jayaraman, A.; Viswanathan, K.; Raman, R.; Sasisekharan, R.; Bennink, J.R. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 2009, 326, 734–736. [Google Scholar] [CrossRef]
- Yewdell, J.W. Antigenic drift: Understanding COVID-19. Immunity 2021, 54, 2681–2687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Schmidt, F.; Muecksch, F.; Wang, Z.; Gazumyan, A.; Nussenzweig, M.C.; Gaebler, C.; Caskey, M.; Hatziioannou, T.; Bieniasz, P.D. SARS-CoV-2 spike glycosylation affects function and neutralization sensitivity. mBio 2024, 15, e01672-23. [Google Scholar] [CrossRef]
- Corti, D.; Lanzavecchia, A. Broadly neutralizing antiviral antibodies. Annu. Rev. Immunol. 2013, 31, 705–742. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, G.F. “Breathing” hemagglutinin reveals cryptic epitopes for universal influenza vaccine de-sign. Cell 2019, 177, 1086–1088. [Google Scholar] [CrossRef]
- Gruell, H.; Vanshylla, K.; Weber, T.; Barnes, C.O.; Kreer, C.; Klein, F. Antibody-mediated neutralization of SARS-CoV-2. Immunity 2022, 55, 925–944. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.-H.; Chen, W.-Y.; Su, S.-C.; Lin, H.-T.; Ke, F.-Y.; Liang, K.-H.; Hsu, F.-F.; Kumari, M.; Fu, C.-Y.; Wu, H.-C. Monoclonal antibodies against S2 subunit of spike protein exhibit broad reactivity toward SARS-CoV-2 variants. J. Biomed. Sci. 2022, 29, 108. [Google Scholar] [CrossRef]
- Tsugawa, Y.; Furukawa, K.; Ise, T.; Takayama, M.; Ota, T.; Kuroda, T.; Shano, S.; Hashimoto, T.; Konishi, H.; Ishihara, T. Discovery of anti-SARS-CoV-2 S2 protein antibody CV804 with broad-spectrum reactivity with various beta coronaviruses and analysis of its pharmacological properties in vitro and in vivo. PLoS ONE 2024, 19, e0300297. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, M.; Saied, A.A.; Mitra, S.; Alhumaydhi, F.A.; Emran, T.B.; Wilairatana, P. Omicron variant (B. 1.1. 529) and its sublineages: What do we know so far amid the emergence of recombinant variants of SARS-CoV-2? Biomed. Pharmacother. 2022, 154, 113522. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Prime, Schrödinger, LLC, New York, NY, USA. 2022. Available online: https://www.schrodinger.com/platform/products/prime/ (accessed on 20 December 2024).
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. Available online: https://jmlr.org/papers/v12/pedregosa11a.html (accessed on 20 December 2024).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | Number of H-Bonds | Number of Contacts | Buried Surface Area (nm2) | KD Values (nM) * | References |
|---|---|---|---|---|---|
| Wild Type | 6.02 | 263.39 | 17.73 | 24.4, 21.9, 16.4 | [43,44,45] |
| Delta | 7.57 | 316.28 | 18.42 | 25.1, 13.5, 2.85 | [43,44,46] |
| BA.1 | 9.99 | 276.97 | 18.17 | 19.5, 14.5, 9.74, 8.85 | [43,44,45,46] |
| BA.1.1 | 5.49 | 293.07 | 18.31 | 5.9 | [43] |
| BA.2 | 8.24 | 312.61 | 18.98 | 10.0, 10.8, 2.99 | [43,44,45] |
| BA.3 | 9.22 | 313.23 | 19.10 | 22.1, 26.5 | [43,44] |
| BA.4/5 | 4.97 | 274.95 | 17.51 | 14.4, 3.1, 10.7 | [44,45,47] |
| BF.7 | 4.57 | 263.06 | 17.59 | 12.97 | [47] |
| XBB.1.16 | 7.38 | 285.76 | 18.53 | 3.2 | [45] |
| XBB.1.9.1 | 5.35 | 286.20 | 17.89 | 3.5, 13.90, 6.0 | [45,47,48] |
| JN.1 | 7.00 | 283.58 | 19.37 | 13 | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, C.; Lupala, C.S.; Wang, D.; Li, X.; Tang, L.-H.; Li, X. Structural and Energetic Insights into SARS-CoV-2 Evolution: Analysis of hACE2–RBD Binding in Wild-Type, Delta, and Omicron Subvariants. Int. J. Mol. Sci. 2025, 26, 3776. https://doi.org/10.3390/ijms26083776
Tang C, Lupala CS, Wang D, Li X, Tang L-H, Li X. Structural and Energetic Insights into SARS-CoV-2 Evolution: Analysis of hACE2–RBD Binding in Wild-Type, Delta, and Omicron Subvariants. International Journal of Molecular Sciences. 2025; 26(8):3776. https://doi.org/10.3390/ijms26083776
Chicago/Turabian StyleTang, Can, Cecylia S. Lupala, Ding Wang, Xiangcheng Li, Lei-Han Tang, and Xuefei Li. 2025. "Structural and Energetic Insights into SARS-CoV-2 Evolution: Analysis of hACE2–RBD Binding in Wild-Type, Delta, and Omicron Subvariants" International Journal of Molecular Sciences 26, no. 8: 3776. https://doi.org/10.3390/ijms26083776
APA StyleTang, C., Lupala, C. S., Wang, D., Li, X., Tang, L.-H., & Li, X. (2025). Structural and Energetic Insights into SARS-CoV-2 Evolution: Analysis of hACE2–RBD Binding in Wild-Type, Delta, and Omicron Subvariants. International Journal of Molecular Sciences, 26(8), 3776. https://doi.org/10.3390/ijms26083776