
Neuroinflammation and Amyotrophic Lateral Sclerosis: Recent Advances in Anti-Inflammatory Cytokines as Therapeutic Strategies

 and
and
Abstract
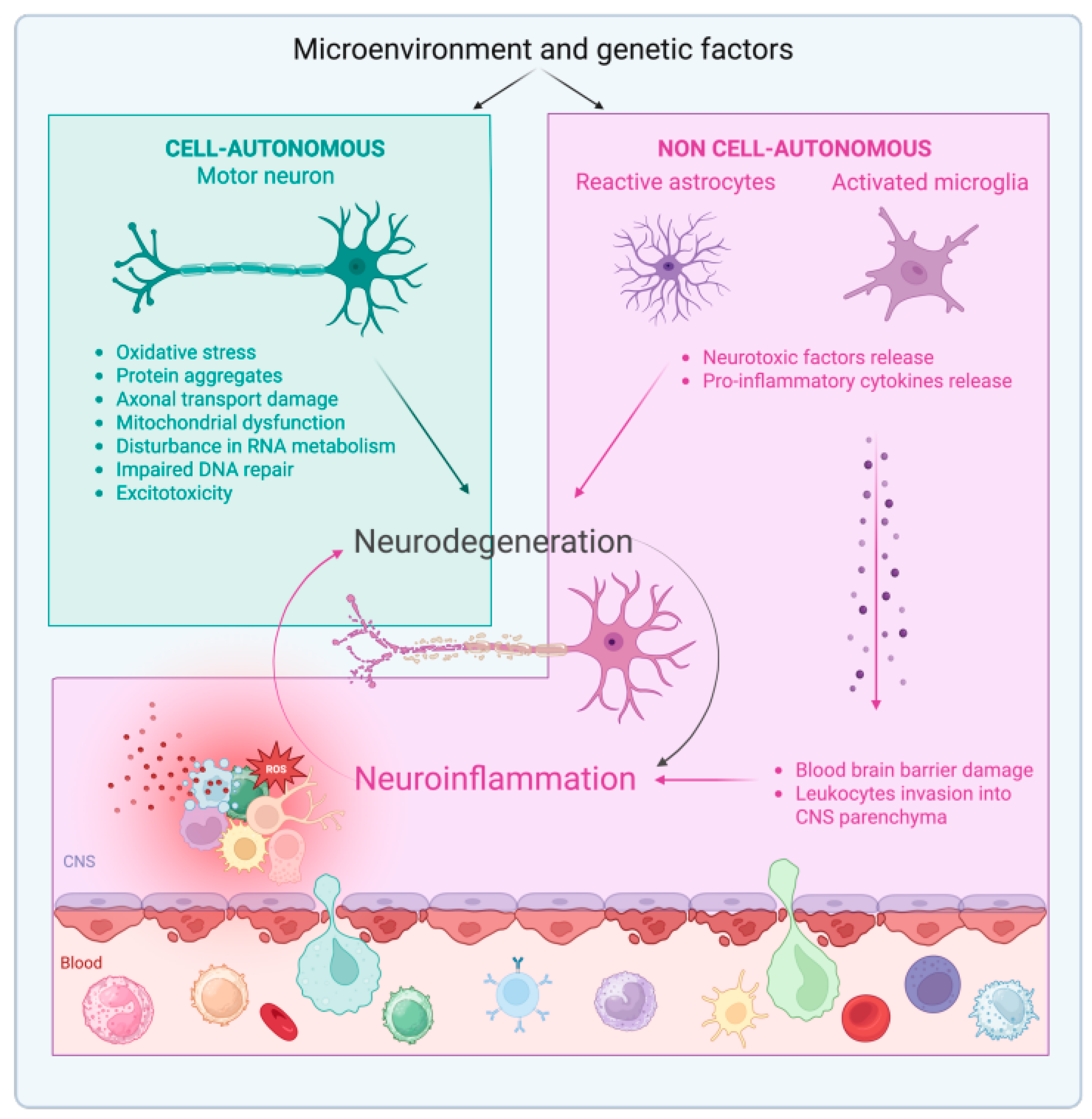
:1. Introduction
Neuroinflammation in ALS
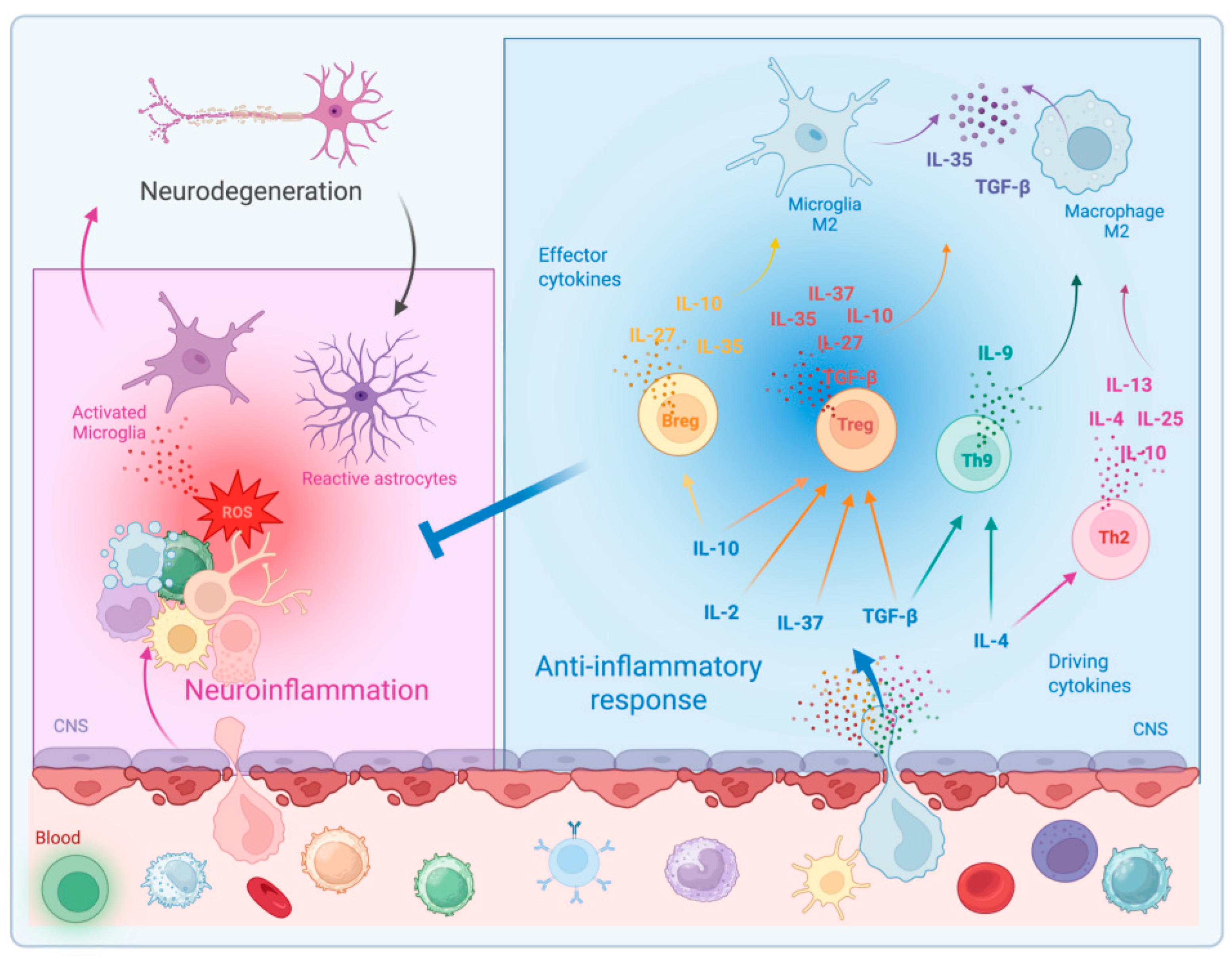
2. Anti-Inflammatory Cytokines in the CNS
2.1. Interleukin-10 in the CNS and in ALS
2.2. Transforming Growth Factor-β in the CNS and in ALS
2.3. Interleukin-4 in the CNS and in ALS
2.4. Interleukin-13 in the CNS and in ALS
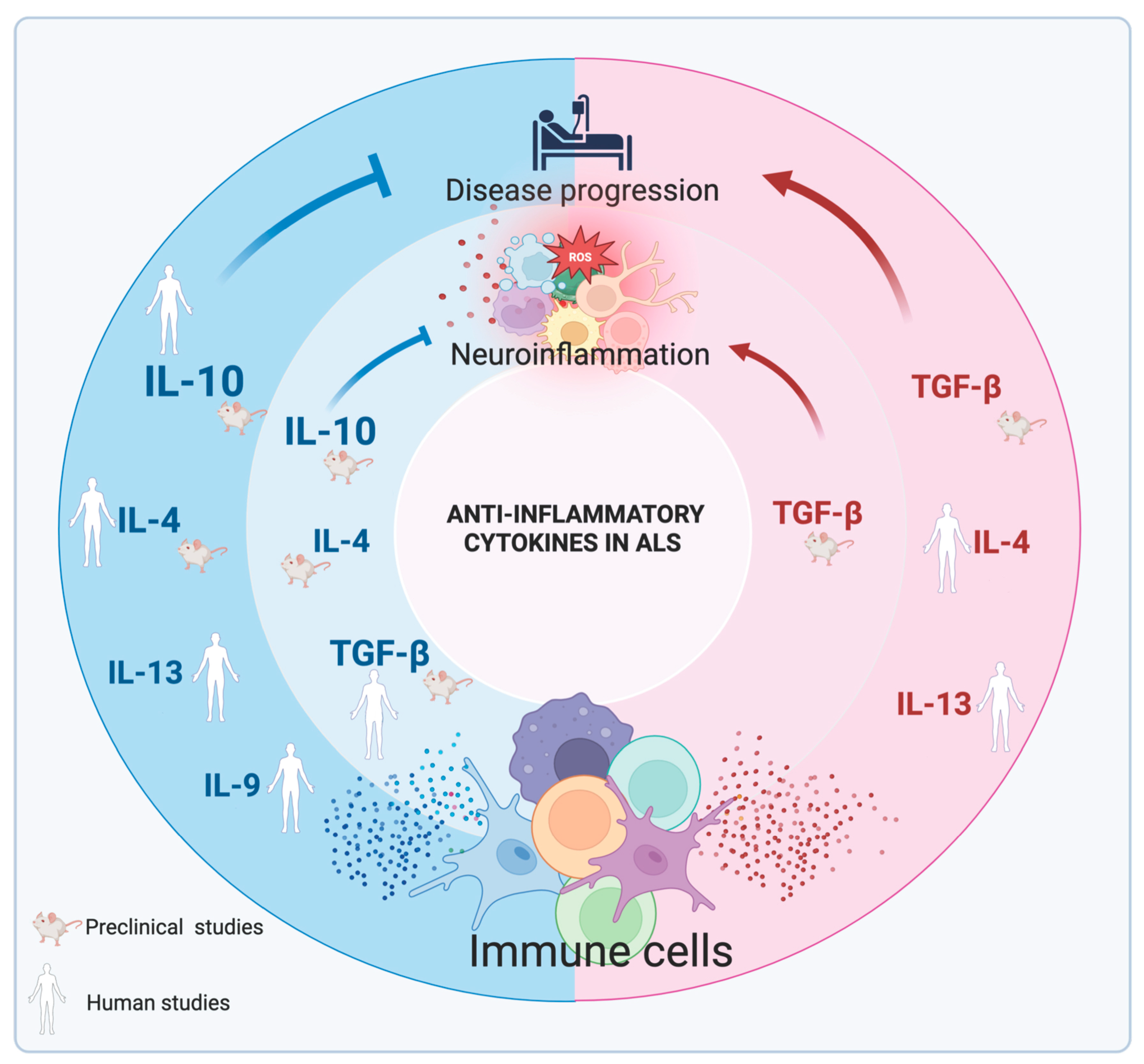
2.5. Interleukin-9 in the CNS and in ALS
2.6. Emerging Anti-Inflammatory Cytokines and Their Role in the CNS
3. Anti-Inflammatory Therapeutic Approaches in ALS

{kind=link}
{kind=link}
{kind=link}
| Name | Functional Role | Clinical Trial | Phase Trial | Clinical Results | Reference |
|---|---|---|---|---|---|
| Anakinra | Antagonist of IL1Ra | NCT01277315 | Phase 2 | No significant reduction of ALSFRS-R | [165] |
| Masitinib | Tyrosine kinase inhibitor | NCT02588677 | Phase 2/3 | Significant decline of ALSFRS-R | [167] |
| Tocilizumab | Neutralizes IL-6R | - | Pilot study | Decrease inflammation cytokines in PBMC | [168] |
| Ibudilast | Inhibitor of toll like receptor 4 and phosphodiesterase 3 and 4 | NCT02714036 | Phase 1 | No significant reductions in motor cortical glial activation measured by PBR28-PET SUVR or CNS neuroaxonal loss, measured by serum NfL | [169] |
| Autologous Treg cells | Induction of suppressive immune response | - | Phase 1 | Reduction of Appel ALS scale for each patient | [170] |
| Low dose IL-2 | Expansion of Treg cells | NCT02059759 | Phase 2 | Significant increase in Treg cells | [174] |
| Rapamycin | Expansion of Treg cells | - | Pilot study | Treatment is safe | [178] |
| Rapamycin | Expansion of Treg cells | NCT03359538 | Phase 2 | Significant increase in Treg cells | [179] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Chakraborty, A.; Diwan, A. Biomarkers and molecular mechanisms of Amyotrophic Lateral Sclerosis. AIMS Neurosci. 2022, 9, 423–443. [Google Scholar] [CrossRef]
- Waisman, A.; Liblau, R.S.; Becher, B. Innate and adaptive immune responses in the CNS. Lancet Neurol. 2015, 14, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Shechter, R.; London, A.; Schwartz, M. Orchestrated leukocyte recruitment to immune-privileged sites: Absolute barriers versus educational gates. Nat. Rev. Immunol. 2013, 13, 206–218. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018, 25, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front. Cell Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef]
- Troost, D.; Van den Oord, J.J.; Vianney de Jong, J.M. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 1990, 16, 401–410. [Google Scholar] [CrossRef]
- Gille, B.; De Schaepdryver, M.; Dedeene, L.; Goossens, J.; Claeys, K.G.; Van Den Bosch, L.; Tournoy, J.; Van Damme, P.; Poesen, K. Inflammatory markers in cerebrospinal fluid: Independent prognostic biomarkers in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2019, 90, 1338–1346. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, S.; Garbelli, S.; Pasini, A.; Alimonti, D.; Perotti, C.; Melazzini, M.; Bendotti, C.; Mora, G. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J. Neuroimmunol. 2009, 210, 73–79. [Google Scholar] [CrossRef]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion with Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabe, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- McCauley, M.E.; O’Rourke, J.G.; Yanez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.; et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Murdock, B.J.; Famie, J.P.; Piecuch, C.E.; Pawlowski, K.D.; Mendelson, F.E.; Pieroni, C.H.; Iniguez, S.D.; Zhao, L.; Goutman, S.A.; Feldman, E.L. NK cells associate with ALS in a sex- and age-dependent manner. JCI Insight 2021, 6, e147129. [Google Scholar] [CrossRef]
- Murdock, B.J.; Zhou, T.; Kashlan, S.R.; Little, R.J.; Goutman, S.A.; Feldman, E.L. Correlation of Peripheral Immunity with Rapid Amyotrophic Lateral Sclerosis Progression. JAMA Neurol. 2017, 74, 1446–1454. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhao, W.; Beers, D.R.; Yen, A.A.; Xie, W.; Henkel, J.S.; Appel, S.H. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J. Neurochem. 2007, 102, 2008–2019. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5. [Google Scholar] [CrossRef]
- Markovinovic, A.; Ljutic, T.; Beland, L.C.; Munitic, I. Optineurin Insufficiency Disbalances Proinflammatory and Anti-inflammatory Factors by Reducing Microglial IFN-beta Responses. Neuroscience 2018, 388, 139–151. [Google Scholar] [CrossRef]
- Levings, M.K.; Bacchetta, R.; Schulz, U.; Roncarolo, M.G. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int. Arch. Allergy Immunol. 2002, 129, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, K.; Onodera, A.; Kiuchi, M.; Tsuji, K.; Hirahara, K.; Nakayama, T. Conventional and pathogenic Th2 cells in inflammation, tissue repair, and fibrosis. Front. Immunol. 2022, 13, 945063. [Google Scholar] [CrossRef]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Ohm, M.; Geffers, R.; Hiller, K.; Dinarello, C.A.; Korte, M. IL-37 expression reduces acute and chronic neuroinflammation and rescues cognitive impairment in an Alzheimer’s disease mouse model. Elife 2022, 11, e75889. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.S.; Keating, B.A.; Perera, C.J.; Lees, J.G.; Tonkin, R.S.; Makker, P.G.S.; Carrive, P.; Butovsky, O.; Moalem-Taylor, G. Regulatory T Cells and Their Derived Cytokine, Interleukin-35, Reduce Pain in Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2019, 39, 2326–2346. [Google Scholar] [CrossRef]
- Maiorino, C.; Khorooshi, R.; Ruffini, F.; Lobner, M.; Bergami, A.; Garzetti, L.; Martino, G.; Owens, T.; Furlan, R. Lentiviral-mediated administration of IL-25 in the CNS induces alternative activation of microglia. Gene Ther. 2013, 20, 487–496. [Google Scholar] [CrossRef]
- Casella, G.; Finardi, A.; Descamps, H.; Colombo, F.; Maiorino, C.; Ruffini, F.; Patrone, M.; Degano, M.; Martino, G.; Muzio, L.; et al. IL-27, but not IL-35, inhibits neuroinflammation through modulating GM-CSF expression. Sci. Rep. 2017, 7, 16547. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- de Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; de Vries, J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, G.; Shin, H.J.; Hyun, J.W.; Kim, S.H.; Lee, E.; Kim, H.J. Restoration of regulatory B cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J. NeuroInflamm. 2018, 15, 300. [Google Scholar] [CrossRef]
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Zoppis, M.; Agostini, S.; Longhi, R.; Caputo, D.; Mendozzi, L.; Rovaris, M.; et al. B Lymphocytes in Multiple Sclerosis: Bregs and BTLA/CD272 Expressing-CD19+ Lymphocytes Modulate Disease Severity. Sci. Rep. 2016, 6, 29699. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Das, M.P.; Howard, E.D.; Weiner, H.L.; Sobel, R.A.; Kuchroo, V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998, 161, 3299–3306. [Google Scholar] [CrossRef]
- Bido, S.; Nannoni, M.; Muggeo, S.; Gambare, D.; Ruffini, G.; Bellini, E.; Passeri, L.; Iaia, S.; Luoni, M.; Provinciali, M.; et al. Microglia-specific IL-10 gene delivery inhibits neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Sci. Transl. Med. 2024, 16, eadm8563. [Google Scholar] [CrossRef]
- Brodovitch, A.; Boucraut, J.; Delmont, E.; Parlanti, A.; Grapperon, A.M.; Attarian, S.; Verschueren, A. Combination of serum and CSF neurofilament-light and neuroinflammatory biomarkers to evaluate ALS. Sci. Rep. 2021, 11, 703. [Google Scholar] [CrossRef]
- Guo, J.; Yang, X.; Gao, L.; Zang, D. Evaluating the levels of CSF and serum factors in ALS. Brain Behav. 2017, 7, e00637. [Google Scholar] [CrossRef]
- Su, X.W.; Simmons, Z.; Mitchell, R.M.; Kong, L.; Stephens, H.E.; Connor, J.R. Biomarker-based predictive models for prognosis in amyotrophic lateral sclerosis. JAMA Neurol. 2013, 70, 1505–1511. [Google Scholar] [CrossRef]
- Furukawa, T.; Matsui, N.; Fujita, K.; Nodera, H.; Shimizu, F.; Miyamoto, K.; Takahashi, Y.; Kanda, T.; Kusunoki, S.; Izumi, Y.; et al. CSF cytokine profile distinguishes multifocal motor neuropathy from progressive muscular atrophy. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e138. [Google Scholar] [CrossRef]
- Ayers, J.I.; Fromholt, S.; Sinyavskaya, O.; Siemienski, Z.; Rosario, A.M.; Li, A.; Crosby, K.W.; Cruz, P.E.; DiNunno, N.M.; Janus, C.; et al. Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol. Ther. 2015, 23, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.R.; Ibanez, K.R.; Yaroshenko, M.; Diaz, C.C.; Borchelt, D.R.; Chakrabarty, P. IL-10 based immunomodulation initiated at birth extends lifespan in a familial mouse model of amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 20862. [Google Scholar] [CrossRef]
- Gravel, M.; Beland, L.C.; Soucy, G.; Abdelhamid, E.; Rahimian, R.; Gravel, C.; Kriz, J. IL-10 Controls Early Microglial Phenotypes and Disease Onset in ALS Caused by Misfolded Superoxide Dismutase 1. J. Neurosci. 2016, 36, 1031–1048. [Google Scholar] [CrossRef]
- Fabbrizio, P.; Margotta, C.; D’Agostino, J.; Suanno, G.; Quetti, L.; Bendotti, C.; Nardo, G. Intramuscular IL-10 Administration Enhances the Activity of Myogenic Precursor Cells and Improves Motor Function in ALS Mouse Model. Cells 2023, 12, 1016. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Mirshafiey, A.; Mohsenzadegan, M. TGF-beta as a promising option in the treatment of multiple sclerosis. Neuropharmacology 2009, 56, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Ke, K.F.; Lu, J.H.; Qiu, Y.H.; Peng, Y.P. Protection of TGF-beta1 against neuroinflammation and neurodegeneration in Abeta1-42-induced Alzheimer’s disease model rats. PLoS ONE 2015, 10, e0116549. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupe, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Esmaeilzadeh, A.; Mohammadi, V.; Elahi, R. Transforming growth factor beta (TGF-beta) pathway in the immunopathogenesis of multiple sclerosis (MS); molecular approaches. Mol. Biol. Rep. 2023, 50, 6121–6131. [Google Scholar] [CrossRef]
- Meroni, M.; Crippa, V.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Soraru, G.; et al. Transforming growth factor beta 1 signaling is altered in the spinal cord and muscle of amyotrophic lateral sclerosis mice and patients. Neurobiol. Aging 2019, 82, 48–59. [Google Scholar] [CrossRef]
- Phatnani, H.P.; Guarnieri, P.; Friedman, B.A.; Carrasco, M.A.; Muratet, M.; O’Keeffe, S.; Nwakeze, C.; Pauli-Behn, F.; Newberry, K.M.; Meadows, S.K.; et al. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. USA 2013, 110, E756–E765. [Google Scholar] [CrossRef]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-derived TGF-beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef]
- Peters, S.; Zitzelsperger, E.; Kuespert, S.; Iberl, S.; Heydn, R.; Johannesen, S.; Petri, S.; Aigner, L.; Thal, D.R.; Hermann, A.; et al. The TGF-beta System as a Potential Pathogenic Player in Disease Modulation of Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 669. [Google Scholar] [CrossRef]
- Gonzalez, D.; Cuenca, X.; Allende, M.L. Knockdown of tgfb1a partially improves ALS phenotype in a transient zebrafish model. Front. Cell Neurosci. 2024, 18, 1384085. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Vitti, G.F.; Burgess, D.R.; Whitty, G.A.; Piccoli, D.S.; Hamilton, J.A. Potential antiinflammatory effects of interleukin 4: Suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2. Proc. Natl. Acad. Sci. USA 1989, 86, 3803–3807. [Google Scholar] [CrossRef]
- Fenton, M.J.; Buras, J.A.; Donnelly, R.P. IL-4 reciprocally regulates IL-1 and IL-1 receptor antagonist expression in human monocytes. J. Immunol. 1992, 149, 1283–1288. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science 1993, 261, 472–475. [Google Scholar] [CrossRef]
- Ferber, I.A.; Lee, H.J.; Zonin, F.; Heath, V.; Mui, A.; Arai, N.; O’Garra, A. GATA-3 significantly downregulates IFN-gamma production from developing Th1 cells in addition to inducing IL-4 and IL-5 levels. Clin. Immunol. 1999, 91, 134–144. [Google Scholar] [CrossRef]
- Luzina, I.G.; Keegan, A.D.; Heller, N.M.; Rook, G.A.; Shea-Donohue, T.; Atamas, S.P. Regulation of inflammation by interleukin-4: A review of “alternatives”. J. Leukoc. Biol. 2012, 92, 753–764. [Google Scholar] [CrossRef]
- Heeb, L.E.M.; Egholm, C.; Boyman, O. Evolution and function of interleukin-4 receptor signaling in adaptive immunity and neutrophils. Genes. Immun. 2020, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, X.; Deng, P.; Hui, H.; Chen, B.; An, J.; Zhang, G.; Shi, K.; Wang, J.; He, Y.; et al. Interleukin-4 from curcumin-activated OECs emerges as a central modulator for increasing M2 polarization of microglia/macrophage in OEC anti-inflammatory activity for functional repair of spinal cord injury. Cell Commun. Signal 2024, 22, 162. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ding, S.; Huang, W.; Hu, J.; Huang, S.; Zhang, Y.; Zhuge, Q. Interleukin-4 Ameliorates the Functional Recovery of Intracerebral Hemorrhage Through the Alternative Activation of Microglia/Macrophage. Front. Neurosci. 2016, 10, 61. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Maresz, K.; Tan, Y.; Dittel, B.N. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 2007, 27, 10714–10721. [Google Scholar] [CrossRef]
- Shaw, M.K.; Lorens, J.B.; Dhawan, A.; DalCanto, R.; Tse, H.Y.; Tran, A.B.; Bonpane, C.; Eswaran, S.L.; Brocke, S.; Sarvetnick, N.; et al. Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J. Exp. Med. 1997, 185, 1711–1714. [Google Scholar] [CrossRef]
- Furlan, R.; Poliani, P.L.; Galbiati, F.; Bergami, A.; Grimaldi, L.M.; Comi, G.; Adorini, L.; Martino, G. Central nervous system delivery of interleukin 4 by a nonreplicative herpes simplex type 1 viral vector ameliorates autoimmune demyelination. Hum. Gene Ther. 1998, 9, 2605–2617. [Google Scholar] [CrossRef]
- Vogelaar, C.F.; Mandal, S.; Lerch, S.; Birkner, K.; Birkenstock, J.; Buhler, U.; Schnatz, A.; Raine, C.S.; Bittner, S.; Vogt, J.; et al. Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation. Sci. Transl. Med. 2018, 10, eaao2304. [Google Scholar] [CrossRef]
- Rossi, C.; Cusimano, M.; Zambito, M.; Finardi, A.; Capotondo, A.; Garcia-Manteiga, J.M.; Comi, G.; Furlan, R.; Martino, G.; Muzio, L. Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death Dis. 2018, 9, 250. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Liao, B.; Henkel, J.S.; Appel, S.H. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol. Dis. 2012, 48, 418–428. [Google Scholar] [CrossRef]
- Femiano, C.; Bruno, A.; Gilio, L.; Buttari, F.; Dolcetti, E.; Galifi, G.; Azzolini, F.; Borrelli, A.; Furlan, R.; Finardi, A.; et al. Inflammatory signature in amyotrophic lateral sclerosis predicting disease progression. Sci. Rep. 2024, 14, 19796. [Google Scholar] [CrossRef] [PubMed]
- Minty, A.; Chalon, P.; Derocq, J.M.; Dumont, X.; Guillemot, J.C.; Kaghad, M.; Labit, C.; Leplatois, P.; Liauzun, P.; Miloux, B.; et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature 1993, 362, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M.; Finkelman, F.D. Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci. Signal 2008, 1, pe55. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 2013, 54, 1–16. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, F.; Cheng, H.; Su, M.; Wang, Y. Macrophage polarization: An important role in inflammatory diseases. Front. Immunol. 2024, 15, 1352946. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Hamzei Taj, S.; Le Blon, D.; Hoornaert, C.; Daans, J.; Quarta, A.; Praet, J.; Van der Linden, A.; Ponsaerts, P.; Hoehn, M. Targeted intracerebral delivery of the anti-inflammatory cytokine IL13 promotes alternative activation of both microglia and macrophages after stroke. J. Neuroinflamm. 2018, 15, 174. [Google Scholar] [CrossRef]
- Van Broeckhoven, J.; Erens, C.; Sommer, D.; Scheijen, E.; Sanchez, S.; Vidal, P.M.; Dooley, D.; Van Breedam, E.; Quarta, A.; Ponsaerts, P.; et al. Macrophage-based delivery of interleukin-13 improves functional and histopathological outcomes following spinal cord injury. J. Neuroinflamm. 2022, 19, 102. [Google Scholar] [CrossRef]
- Kolosowska, N.; Keuters, M.H.; Wojciechowski, S.; Keksa-Goldsteine, V.; Laine, M.; Malm, T.; Goldsteins, G.; Koistinaho, J.; Dhungana, H. Peripheral Administration of IL-13 Induces Anti-inflammatory Microglial/Macrophage Responses and Provides Neuroprotection in Ischemic Stroke. Neurotherapeutics 2019, 16, 1304–1319. [Google Scholar] [CrossRef]
- Miao, W.; Zhao, Y.; Huang, Y.; Chen, D.; Luo, C.; Su, W.; Gao, Y. IL-13 Ameliorates Neuroinflammation and Promotes Functional Recovery after Traumatic Brain Injury. J. Immunol. 2020, 204, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Dooley, D.; Lemmens, E.; Vangansewinkel, T.; Le Blon, D.; Hoornaert, C.; Ponsaerts, P.; Hendrix, S. Cell-Based Delivery of Interleukin-13 Directs Alternative Activation of Macrophages Resulting in Improved Functional Outcome after Spinal Cord Injury. Stem. Cell Rep. 2016, 7, 1099–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Olde Heuvel, F.; Rehman, R.; Aousji, O.; Froehlich, A.; Li, Z.; Jark, R.; Zhang, W.; Conquest, A.; Woelfle, S.; et al. Interleukin-13 and its receptor are synaptic proteins involved in plasticity and neuroprotection. Nat. Commun. 2023, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Lee, D.Y.; Park, K.W.; Kim, S.U.; Yang, M.S.; Joe, E.H.; Jin, B.K. Microglia expressing interleukin-13 undergo cell death and contribute to neuronal survival in vivo. Glia 2004, 46, 142–152. [Google Scholar] [CrossRef]
- Yang, M.S.; Park, E.J.; Sohn, S.; Kwon, H.J.; Shin, W.H.; Pyo, H.K.; Jin, B.; Choi, K.S.; Jou, I.; Joe, E.H. Interleukin-13 and -4 induce death of activated microglia. Glia 2002, 38, 273–280. [Google Scholar] [CrossRef]
- Yang, M.S.; Ji, K.A.; Jeon, S.B.; Jin, B.K.; Kim, S.U.; Jou, I.; Joe, E. Interleukin-13 enhances cyclooxygenase-2 expression in activated rat brain microglia: Implications for death of activated microglia. J. Immunol. 2006, 177, 1323–1329. [Google Scholar] [CrossRef]
- Cash, E.; Minty, A.; Ferrara, P.; Caput, D.; Fradelizi, D.; Rott, O. Macrophage-inactivating IL-13 suppresses experimental autoimmune encephalomyelitis in rats. J. Immunol. 1994, 153, 4258–4267. [Google Scholar] [CrossRef]
- Torkildsen, O.; Brunborg, L.A.; Myhr, K.M.; Bo, L. The cuprizone model for demyelination. Acta Neurol. Scand. Suppl. 2008, 188, 72–76. [Google Scholar] [CrossRef]
- Guglielmetti, C.; Le Blon, D.; Santermans, E.; Salas-Perdomo, A.; Daans, J.; De Vocht, N.; Shah, D.; Hoornaert, C.; Praet, J.; Peerlings, J.; et al. Interleukin-13 immune gene therapy prevents CNS inflammation and demyelination via alternative activation of microglia and macrophages. Glia 2016, 64, 2181–2200. [Google Scholar] [CrossRef]
- Azimzadeh, M.; Mahmoodi, M.; Kazemi, M.; Hakemi, M.G.; Jafarinia, M.; Eslami, A.; Salehi, H.; Amirpour, N. The immunoregulatory and neuroprotective effects of human adipose derived stem cells overexpressing IL-11 and IL-13 in the experimental autoimmune encephalomyelitis mice. Int. Immunopharmacol. 2020, 87, 106808. [Google Scholar] [CrossRef]
- Rossi, S.; Mancino, R.; Bergami, A.; Mori, F.; Castelli, M.; De Chiara, V.; Studer, V.; Mataluni, G.; Sancesario, G.; Parisi, V.; et al. Potential role of IL-13 in neuroprotection and cortical excitability regulation in multiple sclerosis. Mult. Scler. 2011, 17, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Kawano, Y.; Tateishi, T.; Kikuchi, H.; Osoegawa, M.; Ohyagi, Y.; Kira, J. Increased IL-13-producing T cells in ALS: Positive correlations with disease severity and progression rate. J. Neuroimmunol. 2007, 182, 232–235. [Google Scholar] [CrossRef]
- Xu, C.Z.; Huan, X.; Luo, S.S.; Zhong, H.H.; Zhao, C.B.; Chen, Y.; Zou, Z.Y.; Chen, S. Serum cytokines profile changes in amyotrophic lateral sclerosis. Heliyon 2024, 10, e28553. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Van Brandwijk, R.; Fischer, H.G.; Rude, E. Establishment of different T cell sublines using either interleukin 2 or interleukin 4 as growth factors. Eur. J. Immunol. 1990, 20, 1709–1715. [Google Scholar] [CrossRef]
- Goswami, R.; Kaplan, M.H. A brief history of IL-9. J. Immunol. 2011, 186, 3283–3288. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef]
- Stassen, M.; Schmitt, E.; Bopp, T. From interleukin-9 to T helper 9 cells. Ann. N. Y. Acad. Sci. 2012, 1247, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, J.B.; Renauld, J.C. Interleukin 9 and its receptor: An overview of structure and function. Int. Rev. Immunol. 1998, 16, 345–364. [Google Scholar] [CrossRef]
- Yin, T.; Keller, S.R.; Quelle, F.W.; Witthuhn, B.A.; Tsang, M.L.; Lienhard, G.E.; Ihle, J.N.; Yang, Y.C. Interleukin-9 induces tyrosine phosphorylation of insulin receptor substrate-1 via JAK tyrosine kinases. J. Biol. Chem. 1995, 270, 20497–20502. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kubatzky, K.F.; Mitra, D.K. An Update on Interleukin-9: From Its Cellular Source and Signal Transduction to Its Role in Immunopathogenesis. Int. J. Mol. Sci. 2019, 20, 2113. [Google Scholar] [CrossRef]
- Noelle, R.J.; Nowak, E.C. Cellular sources and immune functions of interleukin-9. Nat. Rev. Immunol. 2010, 10, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Levitt, R.C.; McLane, M.P.; MacDonald, D.; Ferrante, V.; Weiss, C.; Zhou, T.; Holroyd, K.J.; Nicolaides, N.C. IL-9 pathway in asthma: New therapeutic targets for allergic inflammatory disorders. J. Allergy Clin. Immunol. 1999, 103, S485–S491. [Google Scholar] [CrossRef]
- Doherty, T.A.; Broide, D.H. Insights into the biology of IL-9 in asthma. J. Allergy Clin. Immunol. 2022, 150, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, H.; Humphreys, N.; Renauld, J.C.; Van Snick, J.; Grencis, R. Interleukin-9 is involved in host protective immunity to intestinal nematode infection. Eur. J. Immunol. 1997, 27, 2536–2540. [Google Scholar] [CrossRef]
- Faulkner, H.; Renauld, J.C.; Van Snick, J.; Grencis, R.K. Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect. Immun. 1998, 66, 3832–3840. [Google Scholar] [CrossRef]
- Pilette, C.; Ouadrhiri, Y.; Van Snick, J.; Renauld, J.C.; Staquet, P.; Vaerman, J.P.; Sibille, Y. Oxidative burst in lipopolysaccharide-activated human alveolar macrophages is inhibited by interleukin-9. Eur. Respir. J. 2002, 20, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Pilette, C.; Ouadrhiri, Y.; Van Snick, J.; Renauld, J.C.; Staquet, P.; Vaerman, J.P.; Sibille, Y. IL-9 inhibits oxidative burst and TNF-alpha release in lipopolysaccharide-stimulated human monocytes through TGF-beta. J. Immunol. 2002, 168, 4103–4111. [Google Scholar] [CrossRef]
- Donninelli, G.; Saraf-Sinik, I.; Mazziotti, V.; Capone, A.; Grasso, M.G.; Battistini, L.; Reynolds, R.; Magliozzi, R.; Volpe, E. Interleukin-9 regulates macrophage activation in the progressive multiple sclerosis brain. J. Neuroinflamm. 2020, 17, 149. [Google Scholar] [CrossRef]
- Ruocco, G.; Rossi, S.; Motta, C.; Macchiarulo, G.; Barbieri, F.; De Bardi, M.; Borsellino, G.; Finardi, A.; Grasso, M.G.; Ruggieri, S.; et al. T helper 9 cells induced by plasmacytoid dendritic cells regulate interleukin-17 in multiple sclerosis. Clin. Sci. 2015, 129, 291–303. [Google Scholar] [CrossRef]
- Elyaman, W.; Bradshaw, E.M.; Uyttenhove, C.; Dardalhon, V.; Awasthi, A.; Imitola, J.; Bettelli, E.; Oukka, M.; van Snick, J.; Renauld, J.C.; et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12885–12890. [Google Scholar] [CrossRef]
- Guadalupi, L.; Vanni, V.; Balletta, S.; Caioli, S.; De Vito, F.; Fresegna, D.; Sanna, K.; Nencini, M.; Donninelli, G.; Volpe, E.; et al. Interleukin-9 protects from microglia- and TNF-mediated synaptotoxicity in experimental multiple sclerosis. J. Neuroinflamm. 2024, 21, 128. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Nourbakhsh, B.; Ciric, B.; Zhang, G.X.; Rostami, A. Neutralization of IL-9 ameliorates experimental autoimmune encephalomyelitis by decreasing the effector T cell population. J. Immunol. 2010, 185, 4095–4100. [Google Scholar] [CrossRef] [PubMed]
- Nowak, E.C.; Weaver, C.T.; Turner, H.; Begum-Haque, S.; Becher, B.; Schreiner, B.; Coyle, A.J.; Kasper, L.H.; Noelle, R.J. IL-9 as a mediator of Th17-driven inflammatory disease. J. Exp. Med. 2009, 206, 1653–1660. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef]
- Dixon, K.O.; van der Kooij, S.W.; Vignali, D.A.; van Kooten, C. Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. Eur. J. Immunol. 2015, 45, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Qu, H.; Zeng, Z.; Li, C.; Wan, J. Interleukin-35 from Interleukin-4-Stimulated Macrophages Alleviates Oxygen Glucose Deprivation/Re-oxygenation-Induced Neuronal Cell Death via the Wnt/beta-Catenin Signaling Pathway. Neurotox. Res. 2022, 40, 420–431. [Google Scholar] [CrossRef]
- Huang, A.; Cheng, L.; He, M.; Nie, J.; Wang, J.; Jiang, K. Interleukin-35 on B cell and T cell induction and regulation. J. Inflamm. 2017, 14, 16. [Google Scholar] [CrossRef]
- Liu, G.; Li, M.; Qian, S.; Yu, L.; Qian, L.; Feng, X. Interleukin-35 exhibits protective effects in a rat model of hypoxic-ischemic encephalopathy through the inhibition of microglia-mediated inflammation. Transl. Pediatr. 2022, 11, 651–662. [Google Scholar] [CrossRef]
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef] [PubMed]
- Rickel, E.A.; Siegel, L.A.; Yoon, B.R.; Rottman, J.B.; Kugler, D.G.; Swart, D.A.; Anders, P.M.; Tocker, J.E.; Comeau, M.R.; Budelsky, A.L. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J. Immunol. 2008, 181, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Angkasekwinai, P.; Park, H.; Wang, Y.H.; Wang, Y.H.; Chang, S.H.; Corry, D.B.; Liu, Y.J.; Zhu, Z.; Dong, C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J. Exp. Med. 2007, 204, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakajima, H.; Suzuki, K.; Kagami, S.; Hirose, K.; Suto, A.; Saito, Y.; Iwamoto, I. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood 2003, 101, 3594–3596. [Google Scholar] [CrossRef]
- Kang, C.M.; Jang, A.S.; Ahn, M.H.; Shin, J.A.; Kim, J.H.; Choi, Y.S.; Rhim, T.Y.; Park, C.S. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am. J. Respir. Cell Mol. Biol. 2005, 33, 290–296. [Google Scholar] [CrossRef]
- Wang, Y.H.; Angkasekwinai, P.; Lu, N.; Voo, K.S.; Arima, K.; Hanabuchi, S.; Hippe, A.; Corrigan, C.J.; Dong, C.; Homey, B.; et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J. Exp. Med. 2007, 204, 1837–1847. [Google Scholar] [CrossRef]
- Tamachi, T.; Maezawa, Y.; Ikeda, K.; Kagami, S.; Hatano, M.; Seto, Y.; Suto, A.; Suzuki, K.; Watanabe, N.; Saito, Y.; et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J. Allergy Clin. Immunol. 2006, 118, 606–614. [Google Scholar] [CrossRef]
- Kleinschek, M.A.; Owyang, A.M.; Joyce-Shaikh, B.; Langrish, C.L.; Chen, Y.; Gorman, D.M.; Blumenschein, W.M.; McClanahan, T.; Brombacher, F.; Hurst, S.D.; et al. IL-25 regulates Th17 function in autoimmune inflammation. J. Exp. Med. 2007, 204, 161–170. [Google Scholar] [CrossRef]
- Caruso, R.; Stolfi, C.; Sarra, M.; Rizzo, A.; Fantini, M.C.; Pallone, F.; MacDonald, T.T.; Monteleone, G. Inhibition of monocyte-derived inflammatory cytokines by IL-25 occurs via p38 Map kinase-dependent induction of Socs-3. Blood 2009, 113, 3512–3519. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, C.; Zheng, D.; Wang, Y.; Lee, V.W.; Wang, Y.M.; Zheng, G.; Tan, T.K.; Yu, D.; Alexander, S.I.; et al. IL-25 induces M2 macrophages and reduces renal injury in proteinuric kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front. Immunol. 2021, 12, 696605. [Google Scholar] [CrossRef] [PubMed]
- Shuai, X.; Wei-min, L.; Tong, Y.L.; Dong, N.; Sheng, Z.Y.; Yao, Y.M. Expression of IL-37 contributes to the immunosuppressive property of human CD4+CD25+ regulatory T cells. Sci. Rep. 2015, 5, 14478. [Google Scholar] [CrossRef]
- Rudloff, I.; Cho, S.X.; Lao, J.C.; Ngo, D.; McKenzie, M.; Nold-Petry, C.A.; Nold, M.F. Monocytes and dendritic cells are the primary sources of interleukin 37 in human immune cells. J. Leukoc. Biol. 2017, 101, 901–911. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.R.; Nold, M.F.; Tang, S.C.; Bui, C.B.; Nold, C.A.; Arumugam, T.V.; Drummond, G.R.; Sobey, C.G.; Kim, H.A. IL-37 increases in patients after ischemic stroke and protects from inflammatory brain injury, motor impairment and lung infection in mice. Sci. Rep. 2019, 9, 6922. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, A.; Zandee, S.; Amo-Aparicio, J.; Charabati, M.; Prat, A.; Garlanda, C.; Eisenmesser, E.Z.; Dinarello, C.A.; Lopez-Vales, R. IL-37 exerts therapeutic effects in experimental autoimmune encephalomyelitis through the receptor complex IL-1R5/IL-1R8. Theranostics 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Yazdani, R.; Naziri, H.; Azizi, G.; Ciric, B.; Askari, M.; Ahmadi, A.M.; Aseervatham, J.; Zhang, G.X.; Rostami, A. IL-37 suppresses CNS autoimmunity by increasing the frequency of Treg cells and reducing CD4 + T cell-derived IL-10 production. J. Neuroinflamm. 2024, 21, 301. [Google Scholar] [CrossRef]
- Pflanz, S.; Timans, J.C.; Cheung, J.; Rosales, R.; Kanzler, H.; Gilbert, J.; Hibbert, L.; Churakova, T.; Travis, M.; Vaisberg, E.; et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 2002, 16, 779–790. [Google Scholar] [CrossRef]
- Pflanz, S.; Hibbert, L.; Mattson, J.; Rosales, R.; Vaisberg, E.; Bazan, J.F.; Phillips, J.H.; McClanahan, T.K.; de Waal Malefyt, R.; Kastelein, R.A. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol. 2004, 172, 2225–2231. [Google Scholar] [CrossRef]
- Hibbert, L.; Pflanz, S.; De Waal Malefyt, R.; Kastelein, R.A. IL-27 and IFN-alpha signal via Stat1 and Stat3 and induce T-Bet and IL-12Rbeta2 in naive T cells. J. Interferon Cytokine Res. 2003, 23, 513–522. [Google Scholar] [CrossRef]
- Carl, J.W.; Bai, X.F. IL27: Its roles in the induction and inhibition of inflammation. Int. J. Clin. Exp. Pathol. 2008, 1, 117–123. [Google Scholar]
- Yoshimura, T.; Takeda, A.; Hamano, S.; Miyazaki, Y.; Kinjyo, I.; Ishibashi, T.; Yoshimura, A.; Yoshida, H. Two-sided roles of IL-27: Induction of Th1 differentiation on naive CD4+ T cells versus suppression of proinflammatory cytokine production including IL-23-induced IL-17 on activated CD4+ T cells partially through STAT3-dependent mechanism. J. Immunol. 2006, 177, 5377–5385. [Google Scholar] [CrossRef]
- Batten, M.; Li, J.; Yi, S.; Kljavin, N.M.; Danilenko, D.M.; Lucas, S.; Lee, J.; de Sauvage, F.J.; Ghilardi, N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat. Immunol. 2006, 7, 929–936. [Google Scholar] [CrossRef]
- Stumhofer, J.S.; Laurence, A.; Wilson, E.H.; Huang, E.; Tato, C.M.; Johnson, L.M.; Villarino, A.V.; Huang, Q.; Yoshimura, A.; Sehy, D.; et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat. Immunol. 2006, 7, 937–945. [Google Scholar] [CrossRef]
- Fitzgerald, D.C.; Zhang, G.X.; El-Behi, M.; Fonseca-Kelly, Z.; Li, H.; Yu, S.; Saris, C.J.; Gran, B.; Ciric, B.; Rostami, A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat. Immunol. 2007, 8, 1372–1379. [Google Scholar] [CrossRef]
- Aldridge, D.L.; Moodley, D.; Park, J.; Phan, A.T.; Rausch, M.; White, K.F.; Ren, Y.; Golin, K.; Radaelli, E.; Kedl, R.; et al. Endogenous IL-27 during toxoplasmosis limits early monocyte responses and their inflammatory activation by pathological T cells. mBio 2024, 15, e0008324. [Google Scholar] [CrossRef]
- Wu, X.Z.; Shi, X.Y.; Zhai, K.; Yi, F.S.; Wang, Z.; Wang, W.; Pei, X.B.; Xu, L.L.; Wang, Z.; Shi, H.Z. Activated naive B cells promote development of malignant pleural effusion by differential regulation of T(H)1 and T(H)17 response. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L443–L455. [Google Scholar] [CrossRef]
- Lin, C.H.; Wu, C.J.; Cho, S.; Patkar, R.; Huth, W.J.; Lin, L.L.; Chen, M.C.; Israelsson, E.; Betts, J.; Niedzielska, M.; et al. Selective IL-27 production by intestinal regulatory T cells permits gut-specific regulation of T(H)17 cell immunity. Nat. Immunol. 2023, 24, 2108–2120. [Google Scholar] [CrossRef]
- Choi, J.K.; Yu, C.R.; Bing, S.J.; Jittayasothorn, Y.; Mattapallil, M.J.; Kang, M.; Park, S.B.; Lee, H.S.; Dong, L.; Shi, G.; et al. IL-27-producing B-1a cells suppress neuroinflammation and CNS autoimmune diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2109548118. [Google Scholar] [CrossRef]
- Senecal, V.; Deblois, G.; Beauseigle, D.; Schneider, R.; Brandenburg, J.; Newcombe, J.; Moore, C.S.; Prat, A.; Antel, J.; Arbour, N. Production of IL-27 in multiple sclerosis lesions by astrocytes and myeloid cells: Modulation of local immune responses. Glia 2016, 64, 553–569. [Google Scholar] [CrossRef]
- Lalive, P.H.; Kreutzfeldt, M.; Devergne, O.; Metz, I.; Bruck, W.; Merkler, D.; Pot, C. Increased interleukin-27 cytokine expression in the central nervous system of multiple sclerosis patients. J. Neuroinflamm. 2017, 14, 144. [Google Scholar] [CrossRef]
- Fitzgerald, D.C.; Ciric, B.; Touil, T.; Harle, H.; Grammatikopolou, J.; Das Sarma, J.; Gran, B.; Zhang, G.X.; Rostami, A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 3268–3275. [Google Scholar] [CrossRef]
- Mertens, M.; Singh, J.A. Anakinra for rheumatoid arthritis: A systematic review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef]
- Maier, A.; Deigendesch, N.; Muller, K.; Weishaupt, J.H.; Krannich, A.; Rohle, R.; Meissner, F.; Molawi, K.; Munch, C.; Holm, T.; et al. Interleukin-1 Antagonist Anakinra in Amyotrophic Lateral Sclerosis—A Pilot Study. PLoS ONE 2015, 10, e0139684. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Nunez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Diaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernandez, M.; Marin, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Frontotemporal Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Mizwicki, M.T.; Fiala, M.; Magpantay, L.; Aziz, N.; Sayre, J.; Liu, G.; Siani, A.; Chan, D.; Martinez-Maza, O.; Chattopadhyay, M.; et al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am. J. Neurodegener. Dis. 2012, 1, 305–315. [Google Scholar]
- Babu, S.; Hightower, B.G.; Chan, J.; Zurcher, N.R.; Kivisakk, P.; Tseng, C.J.; Sanders, D.L.; Robichaud, A.; Banno, H.; Evora, A.; et al. Ibudilast (MN-166) in amyotrophic lateral sclerosis- an open label, safety and pharmacodynamic trial. Neuroimage Clin. 2021, 30, 102672. [Google Scholar] [CrossRef]
- Thonhoff, J.R.; Beers, D.R.; Zhao, W.; Pleitez, M.; Simpson, E.P.; Berry, J.D.; Cudkowicz, M.E.; Appel, S.H. Expanded autologous regulatory T-lymphocyte infusions in ALS: A phase I, first-in-human study. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e465. [Google Scholar] [CrossRef]
- Malek, T.R.; Bayer, A.L. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 2004, 4, 665–674. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef]
- Tang, Q.; Adams, J.Y.; Penaranda, C.; Melli, K.; Piaggio, E.; Sgouroudis, E.; Piccirillo, C.A.; Salomon, B.L.; Bluestone, J.A. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008, 28, 687–697. [Google Scholar] [CrossRef]
- Camu, W.; Mickunas, M.; Veyrune, J.L.; Payan, C.; Garlanda, C.; Locati, M.; Juntas-Morales, R.; Pageot, N.; Malaspina, A.; Andreasson, U.; et al. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): A phase 2a randomised, double-blind, placebo-controlled trial. EBioMedicine 2020, 59, 102844. [Google Scholar] [CrossRef]
- Giovannelli, I.; Bayatti, N.; Brown, A.; Wang, D.; Mickunas, M.; Camu, W.; Veyrune, J.L.; Payan, C.; Garlanda, C.; Locati, M.; et al. Amyotrophic lateral sclerosis transcriptomics reveals immunological effects of low-dose interleukin-2. Brain Commun. 2021, 3, fcab141. [Google Scholar] [CrossRef]
- Alarcan, H.; Bruno, C.; Emond, P.; Raoul, C.; Vourc’h, P.; Corcia, P.; Camu, W.; Veyrune, J.L.; Garlanda, C.; Locati, M.; et al. Pharmacometabolomics applied to low-dose interleukin-2 treatment in amyotrophic lateral sclerosis. Ann. N. Y. Acad. Sci. 2024, 1536, 82–91. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chio, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Med. Baltim. 2018, 97, e11119. [Google Scholar] [CrossRef]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; De Biasi, S.; Banchelli, F.; Martinelli, I.; Simonini, C.; Lo Tartaro, D.; Vicini, R.; Fini, N.; et al. Randomized, double-blind, placebo-controlled trial of rapamycin in amyotrophic lateral sclerosis. Nat. Commun. 2023, 14, 4970. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stacchiotti, C.; Mazzella di Regnella, S.; Cinotti, M.; Spalloni, A.; Volpe, E. Neuroinflammation and Amyotrophic Lateral Sclerosis: Recent Advances in Anti-Inflammatory Cytokines as Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 3854. https://doi.org/10.3390/ijms26083854
Stacchiotti C, Mazzella di Regnella S, Cinotti M, Spalloni A, Volpe E. Neuroinflammation and Amyotrophic Lateral Sclerosis: Recent Advances in Anti-Inflammatory Cytokines as Therapeutic Strategies. International Journal of Molecular Sciences. 2025; 26(8):3854. https://doi.org/10.3390/ijms26083854
Chicago/Turabian StyleStacchiotti, Costanza, Simona Mazzella di Regnella, Miriam Cinotti, Alida Spalloni, and Elisabetta Volpe. 2025. "Neuroinflammation and Amyotrophic Lateral Sclerosis: Recent Advances in Anti-Inflammatory Cytokines as Therapeutic Strategies" International Journal of Molecular Sciences 26, no. 8: 3854. https://doi.org/10.3390/ijms26083854
APA StyleStacchiotti, C., Mazzella di Regnella, S., Cinotti, M., Spalloni, A., & Volpe, E. (2025). Neuroinflammation and Amyotrophic Lateral Sclerosis: Recent Advances in Anti-Inflammatory Cytokines as Therapeutic Strategies. International Journal of Molecular Sciences, 26(8), 3854. https://doi.org/10.3390/ijms26083854