
New Frontiers in Multiple Sclerosis Treatment: From Targeting Costimulatory Molecules to Bispecific Antibodies

 , , , and
, , , and
Abstract
1. Introduction
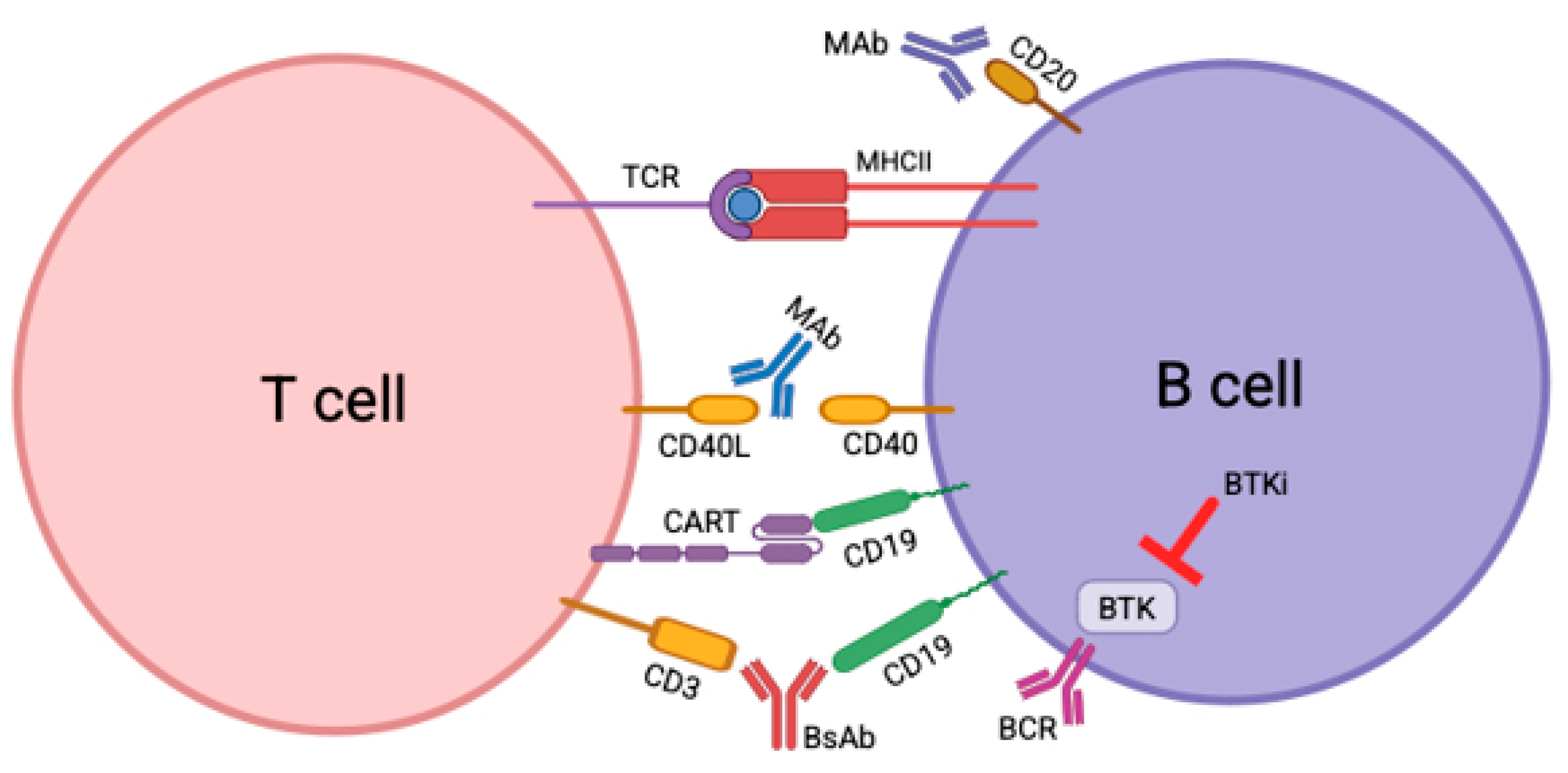
2. CD40/CD40L
2.1. Preclinical Evidence of Target Validation
2.2. Clinical Experience with Anti-CD40L
2.3. Potential Safety Concerns
2.4. Second Generation of CD40/CD40L Therapies in the Pipeline

{kind=link}
| Drug | Trial Phase | Trial Name or Number of Patients | Sponsor | MS Subtype | Recruitment Status | Primary Outcome | Outcome Met? | Secondary Outcome | Outcome Met? | NCT # |
|---|---|---|---|---|---|---|---|---|---|---|
| Toralizumab | I | 12 | Investigator-initiated/Biogen Idec Inc., Cambridge, MA, USA | RRMS | Completed | Adverse events up to 5 years after infusion Serum concentration of drug and anti-drug antibodies up to 18 weeks after infusion Immunologic analysis | Yes | Annualized relapse rate Total brain lesion volume Number of lesions at time of therapy induction and at 2 and 12 weeks post-therapy Total lesion load Maximum tolerated dose | Yes | N/A |
| Frexalimab | II | 129 | Sanofi, Paris, France | RRMS | Active, not recruiting | Number of new Gd-enhancing T1-hyperintense lesions at week 12 | Ongoing | Number of new or enlarging T2 lesions at week 12 Total number of Gd-enhancing T1-hyperintense lesions at week 12 Adverse and serious adverse events until week 316 Anti-drug antibodies until week 316 Maximum concentration of drug until week 316 Time to maximum concentration of drug until week 316 Area under the curve over the dosing interval until week 316 Elimination half-life until week 316 | Ongoing | NCT04879628 |
| Frexalimab | III | FREVIVA:800 | Sanofi, Paris, France | SPMS | Recruiting | Time-to-onset of composite confirmed disability progression up to 6 months after infusion | Ongoing | Time-to-onset of composite confirmed disability progression confirmed over 3 months up to week 204 after infusion Time-to-onset of individual components of the composite confirmed over 3 or 6 months up to week 204 after infusion Time-to-onset of confirmed disability improvement up to week 204 after infusion Number of new and/or enlarging T2 hyperintense lesions per MRI scan up to week 204 after infusion Percent change in brain volume loss as detected by MRI scans from week 24 in comparison to week 204 after infusion Change in cognitive function at week 204 in comparison to baseline as assessed by symbol digit modalities test Change from baseline in multiple sclerosis impact scale 29 version 2 questionnaire scores up to week 204 after infusion Change from baseline in patient reported outcome measurement information system fatigue multiple sclerosis-8a up to week 204 after infusion Annualized relapse rate Number of participants with adverse events up to week 204 after infusion Number of participants with potentially clinically significant abnormalities in lab tests, ECG, and vital signs up to week 204 after infusion Number of participants with antibody over time up to week 204 after infusion Change from baseline in serum immunoglobulin levels up to week 204 after infusion Change from baseline in plasma neurofilament light chain levels up to week 204 after infusion Drug plasma concentration up to week 204 after infusion | Ongoing | NCT06141486 |
| Frexalimab | III | FREXALT: 700 | Sanofi, Paris, France | RRMS | Recruiting | Annualized relapse rate | Ongoing | Time-to-onset of composite confirmed disability worsening over 3 and 6 months up to week 156 after infusion Time-to-onset of composite confirmed disability worsening over 3 or 6 months up to week 156 after infusion Time-to-onset of confirmed disability improvement up to week 156 after infusion Progression independent of relapse activity defined as the time-to-onset of 6 month composite confirmed disability worsening Total number of new and/or enlarging T2 hyperintense lesions as detected by MRI up to week 156 after infusion Total number of new Gd-enhancing T1 hyperintense lesions per scan as detected by MRI up to week 156 after infusion Percent change in brain volume loss as detected by brain MRI scans at the week 156 in comparison to week 24 Change in cognitive function at week 156 compared to baseline as assessed by the symbol digit modalities test Change from baseline in multiple sclerosis impact scale 29 version 2 questionnaire scores over time up to week 156 after infusion Change from baseline in patient-reported outcome measurement information system fatigue MS-8 up to week 156 after infusion Number of participants with adverse events leading to permanent study intervention discontinuation, AESIs, and safety scales up to week 168 after infusion Number of participants with potentially clinically significant abnormalities in lab tests, ECG, and vital signs up to week 168 after infusion Number of participants with anti-drug antibodies up to week 156 after infusion Change from baseline in plasma neurofilament light chain levels up to week 144 after infusion Drug plasma concentration up to week 144 after infusion | Ongoing | NCT06141473 |
3. CAR-T
3.1. Preclinical Evidence of Target Validation
3.2. Clinical Experience of CAR T Cell Therapy in Cancer
3.3. Clinical Experience of CAR T Cell Therapy in Autoimmune Disease
3.4. Potential Safety Concerns
3.5. CAR T Cell Therapies in the Pipeline for MS
4. BTK Inhibitors
4.1. Preclinical Evidence for Target Evaluation
4.2. Clinical Experience with BTKi
4.3. Safety Concerns
4.4. BTKi in the Pipeline
5. Bispecific Antibodies on the Horizon
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schett, G.; Mackensen, A.; Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 2023, 402, 2034–2044. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Fischbach, F.; Richter, J.; Pfeffer, L.K.; Fehse, B.; Berger, S.C.; Reinhardt, S.; Kuhle, J.; Badbaran, A.; Rathje, K.; Gagelmann, N.; et al. CD19-targeted chimeric antigen receptor T cell therapy in two patients with multiple sclerosis. Med 2024, 5, 550–558.e2. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Ysrraelit, M.C.; Fiol, M.P. Benign Multiple Sclerosis: Does it exist? Curr. Neurol. Neurosci. Rep. 2012, 12, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Aarts, S.; Seijkens, T.T.P.; van Dorst, K.J.F.; Dijkstra, C.D.; Kooij, G.; Lutgens, E. The CD40-CD40L Dyad in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front. Immunol. 2017, 8, 1791. [Google Scholar] [CrossRef]
- Comi, G.; Bar-Or, A.; Lassmann, H.; Uccelli, A.; Hartung, H.P.; Montalban, X.; Sørensen, P.S.; Hohlfeld, R.; Hauser, S.L. Role of B Cells in Multiple Sclerosis and Related Disorders. Ann. Neurol. 2021, 89, 13–23. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef]
- Treabă, C.A.; Bălaşa, R.; Podeanu, D.M.; Simu, I.P.; Buruian, M.M. Cerebral lesions of multiple sclerosis: Is gadolinium always irreplaceable in assessing lesion activity? Diagn. Interv. Radiol. 2014, 20, 178–184. [Google Scholar] [CrossRef]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef]
- Edwards Jonathan, C.W.; Szczepański, L.; Szechiński, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close David, R.; Stevens Randall, M.; Shaw, T. Efficacy of B-Cell–Targeted Therapy with Rituximab in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Hauser Stephen, L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan Kottil, W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Steinman, L.; Fox, E.; Hartung, H.P.; Alvarez, E.; Qian, P.; Wray, S.; Robertson, D.; Huang, D.; Selmaj, K.; Wynn, D.; et al. Ublituximab versus Teriflunomide in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2022, 387, 704–714. [Google Scholar] [CrossRef]
- Anolik, J.H.; Barnard, J.; Owen, T.; Zheng, B.; Kemshetti, S.; Looney, R.J.; Sanz, I. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007, 56, 3044–3056. [Google Scholar] [CrossRef]
- Chen, D.; Ireland, S.J.; Remington, G.; Alvarez, E.; Racke, M.K.; Greenberg, B.; Frohman, E.M.; Monson, N.L. CD40-Mediated NF-κB Activation in B Cells Is Increased in Multiple Sclerosis and Modulated by Therapeutics. J. Immunol. 2016, 197, 4257–4265. [Google Scholar] [CrossRef]
- Gerritse, K.; Laman, J.D.; Noelle, R.J.; Aruffo, A.; Ledbetter, J.A.; Boersma, W.J.; Claassen, E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 1996, 93, 2499–2504. [Google Scholar] [CrossRef]
- Dittel, B.N. CD4 T cells: Balancing the coming and going of autoimmune-mediated inflammation in the CNS. Brain Behav. Immun. 2008, 22, 421–430. [Google Scholar] [CrossRef]
- Ots, H.D.; Tracz, J.A.; Vinokuroff, K.E.; Musto, A.E. CD40-CD40L in Neurological Disease. Int. J. Mol. Sci. 2022, 23, 4115. [Google Scholar] [CrossRef] [PubMed]
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11, eaar6584. [Google Scholar] [CrossRef]
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A.; on behalf of the BG9588 Lupus Nephritis Trial Group. A short course of BG9588 (anti–CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727. [Google Scholar] [CrossRef]
- Mills, E.A.; Begay, J.A.; Fisher, C.; Mao-Draayer, Y. Impact of trial design and patient heterogeneity on the identification of clinically effective therapies for progressive MS. Mult. Scler. 2018, 24, 1795–1807. [Google Scholar] [CrossRef]
- Yang, J.; Hamade, M.; Wu, Q.; Wang, Q.; Axtell, R.; Giri, S.; Mao-Draayer, Y. Current and Future Biomarkers in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 5877. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, Q.; Yang, J.; Martens, J.W.; Mills, E.A.; Saad, A.; Chilukuri, P.; Dowling, C.A.; Mao-Draayer, Y. Elevated sCD40L in Secondary Progressive Multiple Sclerosis in Comparison to Non-progressive Benign and Relapsing Remitting Multiple Sclerosis. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211050712. [Google Scholar] [CrossRef]
- Fadul, C.E.; Mao-Draayer, Y.; Ryan, K.A.; Noelle, R.J.; Wishart, H.A.; Channon, J.Y.; Kasper, I.R.; Oliver, B.; Mielcarz, D.W.; Kasper, L.H. Safety and Immune Effects of Blocking CD40 Ligand in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1096. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Andrews, D.; Colvin, R.B.; Sachs, D.H.; Cosimi, A.B. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat. Med. 2000, 6, 114. [Google Scholar] [CrossRef]
- Langer, F.; Ingersoll, S.B.; Amirkhosravi, A.; Meyer, T.; Siddiqui, F.A.; Ahmad, S.; Walker, J.M.; Amaya, M.; Desai, H.; Francis, J.L. The role of CD40 in CD40L- and antibody-mediated platelet activation. Thromb. Haemost. 2005, 93, 1137–1146. [Google Scholar] [CrossRef]
- Vermersch, P.; Granziera, C.; Mao-Draayer, Y.; Cutter, G.; Kalbus, O.; Staikov, I.; Dufek, M.; Saubadu, S.; Bejuit, R.; Truffinet, P.; et al. Inhibition of CD40L with Frexalimab in Multiple Sclerosis. N. Engl. J. Med. 2024, 390, 589–600. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Giovannoni, G.; Hawkes, C.H.; Lechner-Scott, J.; Levy, M.; Yeh, E.A. Are we ready for CD19-targeted CAR T-cell therapies in MS? Mult. Scler. Relat. Disord. 2023, 70, 104590. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Blazek, M.; Ireland, S.; Ortega, S.; Kong, X.; Meeuwissen, A.; Stowe, A.; Carter, L.; Wang, Y.; Herbst, R.; et al. Single dose of glycoengineered anti-CD19 antibody (MEDI551) disrupts experimental autoimmune encephalomyelitis by inhibiting pathogenic adaptive immune responses in the bone marrow and spinal cord while preserving peripheral regulatory mechanisms. J. Immunol. 2014, 193, 4823–4832. [Google Scholar] [CrossRef]
- Gupta, S.; Simic, M.; Sagan, S.A.; Shepherd, C.; Duecker, J.; Sobel, R.A.; Dandekar, R.; Wu, G.F.; Wu, W.; Pak, J.E.; et al. CAR-T Cell-Mediated B-Cell Depletion in Central Nervous System Autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200080. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, R.; Maier, H.J.; Zhang, J.; Lim, S. Kymriah® (tisagenlecleucel)—An overview of the clinical development journey of the first approved CAR-T therapy. Hum. Vaccin. Immunother. 2023, 19, 2210046. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Frigault, M.J.; Dietrich, J.; Gallagher, K.; Roschewski, M.; Jordan, J.T.; Forst, D.; Plotkin, S.R.; Cook, D.; Casey, K.S.; Lindell, K.A.; et al. Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: A phase 1/2 clinical trial. Blood 2022, 139, 2306–2315. [Google Scholar] [CrossRef]
- Schuster, S.J.; Tam, C.S.; Borchmann, P.; Worel, N.; McGuirk, J.P.; Holte, H.; Waller, E.K.; Jaglowski, S.; Bishop, M.R.; Damon, L.E.; et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-Cell Therapy in Autoimmune Disease—A Case Series with Follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef]
- Arunachalam, A.K.; Grégoire, C.; Coutinho de Oliveira, B.; Melenhorst, J.J. Advancing CAR T-cell therapies: Preclinical insights and clinical translation for hematological malignancies. Blood Rev. 2024, 68, 101241. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Qin, C.; Tian, D.-S.; Zhou, L.-Q.; Shang, K.; Huang, L.; Dong, M.-H.; You, Y.-F.; Xiao, J.; Xiong, Y.; Wang, W.; et al. Anti-BCMA CAR T-cell therapy CT103A in relapsed or refractory AQP4-IgG seropositive neuromyelitis optica spectrum disorders: Phase 1 trial interim results. Signal Transduct. Target. Ther. 2023, 8, 5. [Google Scholar] [CrossRef]
- Burger, J.A. Bruton Tyrosine Kinase Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef]
- Carnero Contentti, E.; Correale, J. Bruton’s tyrosine kinase inhibitors: A promising emerging treatment option for multiple sclerosis. Expert. Opin. Emerg. Drugs 2020, 25, 377–381. [Google Scholar] [CrossRef]
- Ringheim, G.E.; Wampole, M.; Oberoi, K. Bruton’s Tyrosine Kinase (BTK) Inhibitors and Autoimmune Diseases: Making Sense of BTK Inhibitor Specificity Profiles and Recent Clinical Trial Successes and Failures. Front. Immunol. 2021, 12, 662223. [Google Scholar] [CrossRef]
- Gruber, R.C.; Chretien, N.; Dufault, M.R.; Proto, J.; Zhang, M.; LaMorte, M.; Havari, E.; Samad, T.A.; Turner, T.; Chomyk, A.; et al. Central Effects of BTK Inhibition in Neuroinflammation (808). Neurology 2020, 94, 808. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef]
- Montalban, X.; Vermersch, P.; Arnold, D.L.; Bar-Or, A.; Cree, B.A.C.; Cross, A.; Havrdova, E.K.; Kappos, L.; Stuve, O.; Wiendl, H.; et al. Design and Baseline Characteristics of Phase 3, Double-Blind, Randomised Trials Evaluating the Efficacy and Safety of Evobrutinib Versus Teriflunomide in Relapsing Multiple Sclerosis (evolutionRMS 1 and 2). Mult. Scler. Relat. Disord. 2023, 80, 105328. [Google Scholar] [CrossRef]
- Jain, R.W.; Yong, V.W. B cells in central nervous system disease: Diversity, locations and pathophysiology. Nat. Rev. Immunol. 2022, 22, 513–524. [Google Scholar] [CrossRef]
- Yong, H.Y.F.; Yong, V.W. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Aigrot, M.S.; Grenningloh, R.; Stankoff, B.; Lubetzki, C.; Boschert, U.; Zalc, B. Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair. Brain Plast. 2020, 5, 123–133. [Google Scholar] [CrossRef]
- Francesco, M. PRN2246, a potent and selective blood brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Mult. Scler. J. 2017, 23, 427–679. [Google Scholar] [CrossRef]
- Bhargava, P.; Kim, S.; Reyes, A.A.; Grenningloh, R.; Boschert, U.; Absinta, M.; Pardo, C.; Van Zijl, P.; Zhang, J.; Calabresi, P.A. Imaging meningeal inflammation in CNS autoimmunity identifies a therapeutic role for BTK inhibition. Brain 2021, 144, 1396–1408. [Google Scholar] [CrossRef]
- Montalban, X.; Vermersch, P.; Arnold, D.L.; Bar-Or, A.; Cree, B.A.C.; Cross, A.H.; Kubala Havrdova, E.; Kappos, L.; Stuve, O.; Wiendl, H.; et al. Safety and efficacy of evobrutinib in relapsing multiple sclerosis (evolutionRMS1 and evolutionRMS2): Two multicentre, randomised, double-blind, active-controlled, phase 3 trials. Lancet Neurol. 2024, 23, 1119–1132. [Google Scholar] [CrossRef]
- Reich, D.S.; Arnold, D.L.; Vermersch, P.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallström, E.; Zhang, X.; Mareš, M.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Owens, T.D.; Smith, P.F.; Redfern, A.; Xing, Y.; Shu, J.; Karr, D.E.; Hartmann, S.; Francesco, M.R.; Langrish, C.L.; Nunn, P.A.; et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin. Transl. Sci. 2022, 15, 442–450. [Google Scholar] [CrossRef]
- Barr, H.J.; Given, K.S.; McClain, C.R.; Gruber, R.C.; Ofengeim, D.; Macklin, W.B.; Bennett, J.L.; Owens, G.; Hughes, E. Microglial BTK Signaling Regulates Immune-Mediated Cortical Demyelination. Mult. Scler. J. 2021, 27, 15–122. [Google Scholar] [CrossRef]
- Nicolas, O.; Cabanis, M.; Brun, P.; Vitse, O.; Soubayrol, P.; Turner, T.J.; Krupka, E. Tolebrutinib demonstrates cerebrospinal fluid exposure at bioactive levels in a single-ascending dose study in healthy volunteers. Mult. Scler. J. 2023, 29, 18–242. [Google Scholar] [CrossRef]
- Piasecka-Stryczynska, K.; Rejdak, K.; Dyroff, M.; Hyvert, Y.; Holmberg, K.; Mandel, M.; Cunha, C.; Mitchell, D.; Martin, E.; Montalban, X. Concentration of evobrutinib, a BTK inhibitor, in cerebrospinal fluid during treatment of patients with relapsing multiple sclerosis in a phase 2 study. Mult. Scler. Relat. Disord. 2021, 51, 103001. [Google Scholar] [CrossRef]
- ECTRIMS 2024—Late Breaking Oral Presentations. Mult. Scler. J. 2024, 30, 1145–1146. [CrossRef]
- Ghez, D.; Calleja, A.; Protin, C.; Baron, M.; Ledoux, M.P.; Damaj, G.; Dupont, M.; Dreyfus, B.; Ferrant, E.; Herbaux, C.; et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood 2018, 131, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Quartermaine, C.; Ghazi, S.M.; Yasin, A.; Awan, F.T.; Fradley, M.; Wiczer, T.; Kalathoor, S.; Ferdousi, M.; Krishan, S.; Habib, A.; et al. Cardiovascular Toxicities of BTK Inhibitors in Chronic Lymphocytic Leukemia: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol 2023, 5, 570–590. [Google Scholar] [CrossRef]
- Nakhoda, S.; Vistarop, A.; Wang, Y.L. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br. J. Haematol. 2023, 200, 137–149. [Google Scholar] [CrossRef]
- Lionakis, M.S.; Dunleavy, K.; Roschewski, M.; Widemann, B.C.; Butman, J.A.; Schmitz, R.; Yang, Y.; Cole, D.E.; Melani, C.; Higham, C.S.; et al. Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell 2017, 31, 833–843.e5. [Google Scholar] [CrossRef]
- Trabolsi, A.; Arumov, A.; Schatz, J.H. Bispecific antibodies and CAR-T cells: Dueling immunotherapies for large B-cell lymphomas. Blood Cancer J. 2024, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, D.; Kemmerich, K.; Haqqani, A.S.; Farrington, G.K. Engineering and pharmacology of blood-brain barrier-permeable bispecific antibodies. Adv. Pharmacol. 2014, 71, 301–335. [Google Scholar] [CrossRef] [PubMed]
- Amgen. FDA Approves Blincyto (Blinatumomab) in CD19-Positive Philadelphia Chromosome-Negative B-Cell Precursor Acute Lymphoblastic Leukemia (B-ALL) in the Consolidation Phase. 2024. Available online: https://www.amgen.com/newsroom/press-releases/2024/06/fda-approves-blincyto-blinatumomab-in-cd19positive-philadelphia-chromosomenegative-bcell-precursor-acute-lymphoblastic-leukemia-ball-in-the-consolidation-phase (accessed on 2 February 2025).
| Drug | Trial Phase | Sponsor | MS Subtype | Recruitment Status | Primary Outcome | Outcome Met? | Secondary Outcome | Outcome Met? | NCT # |
|---|---|---|---|---|---|---|---|---|---|
| KYV-101 (CD19 CAR T) | I | Kyverna Therapeutics, Emeryville, CA, USA | Non-relapsing, progressive | Recruiting | Frequency of dose-limiting toxicities for each dose level up to 12 months after infusion | Ongoing | Adverse events up to 12 months after infusion Drug concentration in the blood and CSF up to 12 months after infusion Level of B cells in blood up to 12 months after infusion Time from infusion to a change in disability and walking score as measured by EDSS Levels of unique unmatched intrathecal oligoclonal bands up to 6 months after infusion Whole brain and gray matter volume up to 12 months after infusion | Ongoing | NCT06138132 |
| KYV-101 (CD19 CAR T) | I | Kyverna Therapeutics, Emeryville, CA, USA | Progressive | Recruiting | Drug concentration following peak expansion in peripheral blood up to week 48 after infusion Adverse events and dose-limiting toxicities up to week 48 after infusion | Ongoing | Proportion of patients with reduction in CSF OCB and/or normalization of CSF IgG index | Ongoing | NCT06451159 |
| KYV-101 (CD19 CAR T) | II | Kyverna Therapeutics, Emeryville, CA, USA | Progressive | Recruiting | Confirmed disability progression as measured by EDSS | Ongoing | Adverse events up to 2 years after infusion Composite confirmed disability progression up to 12 weeks after infusion Drug levels up to 2 years after infusion Level of B cells in blood up to 2 years after infusion Level of serum cytokines up to 2 years after infusion Percentage of patients that developed anti-KYV-101 antibodies | Ongoing | NCT06384976 |
| Universal BCMA CAR T, universal CD19 CAR T | I | Xuanwu Hospital, Beijing, China | Not listed | Not yet recruiting | Dose-limiting toxicities up to 28 days after infusion Adverse events up to 12 months after infusion | Ongoing | Concentration of drug in peripheral blood up to 3 months after infusion Level of B cells in peripheral blood up to 3 months after infusion Changes in pathogenic antibody titers in peripheral blood or CSF at 1, 3, 6, and 12 months after infusion Annualized relapse rate Changes in Myasthenia Gravis Activities of Daily Living (MG-ADL) score at 1, 3, 6, and 12 months after infusion Changes in Inflammatory Neuropathy Cause and Treatment (INCAT) score at 1, 3, 6, and 12 months after infusion Change in number of Gd-enhancing T1 lesions at 6 and 12 months after infusion Change in number of new or enlarging T2 lesions at 6 and 12 months after infusion | Ongoing | NCT06485232 |
| C-CAR168 (CD20/BCMA-directed CAR T) | I | RenJi Hospital, Shanghai, China | RRMS | Recruiting | Adverse events and dose-limiting toxicities up to 3 years after infusion | Ongoing | Proportion of patients who achieved remission at 6 months after infusion Proportion of patients who achieved remission up to 3 years after infusion Proportion of patients who relapsed up to 3 years after infusion Time to response of first remission Progression-free survival up to 3 years after infusion Proportion of patients who achieved either low or no dose of glucocorticoids and/or immunosuppressants up to 3 years after infusion Maximum plasma concentration of drug up to 3 years after infusion Time to achieve maximum plasma concentration of drug up to 3 years after infusion Duration of drug in peripheral blood up to 3 years after infusion Area under the curve of drug in peripheral blood up to 3 years after infusion Clearance of peripheral blood B cells up to 3 years after infusion Decline of serum immunoglobulin up to 3 years after infusion Elevation of peripheral blood complement up to 3 years after infusion Decline of autoantibodies or other disease specific biomarkers up to 3 years after infusion | Ongoing | NCT06249438 |
| CT103A (BCMA CAR T) | I | Tongji Hospital, Wuhan, China | RRMS | Recruiting | Dose-limiting toxicities up to 28 days after infusion Adverse events up to 2 years after infusion | Ongoing | Changes in concentration of soluble BCMA in peripheral blood after infusion up to 2 years after infusion Changes in pathogenic antibody titers in peripheral blood or CSF up to 2 years after infusion Changes in peripheral blood neurofilament light chain concentrations up to 2 years after infusion Number of BCMA CAR gene copies in peripheral blood and CSF up to 15 years after infusion Concentration of BCMA CAR T cells in peripheral blood up to 28 days after infusion | Ongoing | NCT04561557 |
| YTB323 (CD19 CAR T) | I/II | Novartis Pharmaceuticals, Basel, Switzerland | RRMS | Not yet recruiting | Adverse events up to 2 years after infusion | Ongoing | EDSS score up to 2 years after infusion Short-form health survey to assess quality of life up to 2 years after infusion Timed 25-foot walk up to 2 years after infusion 9-Hole Peg Test up to 2 years after infusion Symbol Digit Modalities Test up to 2 years after infusion Fatigue symptoms and impacts questionnaire up to 2 years after infusion Number of new and enlarging T2 lesions and Gd-enhancing T1 lesions up to 2 years after infusion Maximum plasma concentration of drug up to 2 years after infusion Area under the curve of drug up to 2 years after infusion Time to reach maximum concentration of drug up to 2 years after infusion The last quantifiable plasma concentration of drug up to 2 years after infusion The time of the last quantifiable concentration of drug up to 2 years after infusion Humoral immunogenicity of drug up to 2 years after infusion Cellular immunogenicity of drug up to 2 years after infusion Drug meeting release specifications at or above target dose between day 9 and day 2 | Ongoing | NCT06617793 |
| Drug | Trial Phase | Sponsor | Trial Status | Study name and Total Number of Participants | MS Subtype | Primary Outcome | Outcome Met? | Secondary Outcome | Outcome Met? | NCT # |
|---|---|---|---|---|---|---|---|---|---|---|
| Tolebrutinib | III | Sanofi, Paris, France | Completed | GEMINI 1: 824 GEMINI 2: 762 | RRMS compared to teriflunomide | Annualized relapse rate (ARR) (number of adjudicated MS relapses in a year) | No—Annualized relapse rate was low in the teriflunomide arm in both GEMINI 1 and 2 and no difference was observed between tolebrutinib and teriflunomide. | Time-to-onset of 6-month confirmed disability worsening (CDW) | Yes—tolebrutinib demonstrated clear separation from teriflunomide (29% relative risk reduction) in a population with very low relapse activity. | NCT04410978 (GEMINI 1) NCT04410991 (GEMINI 2) |
| Time-to-onset of 3-month CDW | Yes—tolebrutinib demonstrated similar effects on time to 3-month CDW as 6-month CDW. | |||||||||
| Time-to-onset of 6-month confirmed disability improvement | Yes—there was a numerically higher rate of 6-month confirmed disability improvement in the tolebrutinib arm compared to teriflunomide. | |||||||||
| Total number of new Gd-enhancing T1 brain lesions | The number of Gd-enhancing T1 lesions was higher in the tolebrutinib arm. | |||||||||
| Total number of new/enlarging T2 brain lesions | The number of new/enlarging T2 lesions was similar between both treatment arms | |||||||||
| % change in brain volume | Brain volume loss (BVL) from month 6 to EOS was low for both tolebrutinib and placebo groups | |||||||||
| Tolebrutinib | III | Sanofi, Paris, France | Completed | HERCULES: 869 | nrSPMS | 6-month confirmed disability progression (CDP) [time frame: up to approximately 48 months] | Yes-Tolebrutinib showed a 31% risk reduction in time to 6-month CDP vs. placebo | Time-to-onset of 3-month CDP | Tolebrutinib demonstrated a significant effect on time to 3-month CDP | NCT04411641 (HERCULES) |
| Time-to-onset of 6-month CDI | Proportionally more participants experienced CDI on tolebrutinib vs. placebo | |||||||||
| Total number of new or enlarging T2 lesions | Tolebrutinib significantly lowered the annualized rate of new/enlarging T2 lesions vs. placebo | |||||||||
| % change in brain volume | Brain volume loss (BVL) from month 6 to EOS was low for both tolebrutinib and placebo groups | |||||||||
| Evobrutnib | III | Merck KGaA, Darmstadt, Germany | Completed | EVOLUTION RMS1: 807 EVOLUTIONRMS2: 847 | RMS | Annualized relapse rate | No—Evobrutinib did not show superior efficacy to that of teriflunomide in either study | Number of new T1 gadolinium-enhancing lesions | The total number of T1 gadolinium-enhancing lesions and adjusted mean number per scan were numerically higher for evobrutinib than for teriflunomide in both studies | NCT043380 (EVOLUTION RMS1) NCT04338061 |
| New or enlarging T2 lesions | The total number of new or enlarging T2 lesions was similar between groups in both studies and did not show an increase from week 24 in the evobrutinib group | |||||||||
| Serum NfL concentration at week 12, ng/L | Adjusted geometric mean serum NfL concentrations at week 12 showed no difference between evobrutinib and teriflunomide in evolutionRMS1 and a nominal difference in evolutionRMS2. Prespecified exploratory analysis showed a similar reduction in serum NfL by week 96 from baseline between evobrutinib and teriflunomide in both studies, with no increase in serum NfL after week 24 | |||||||||
| Evobrutinib | II | Merck KGaA, Darmstadt, Germany | Completed | RMS, RRMS | Total number of gadolinium-enhancing T1 lesions | Yes—patients with RMS who received 75 mg of evobrutinib once daily had significantly fewer enhancing lesions during weeks 12 through 24 than those who received placebo | Annualized relapse rate (ARR) | No—no significant difference with placebo for either the 25 mg once-daily or 75 mg twice-daily dose of evobrutinib, nor in the annualized relapse rate or disability progression at any dose | NCT02975349 | |
| Evobrutinib | II | Merck KGaA, Darmstadt, Germany | Completed | RRMS | Annualized relapse rate (ARR) | No—failed to reduce ARR in people with MS | N/A | N/A | NCT04338061 | |
| Fenebrutinib | II | Roche–Genentech, Basel, Switzerland | Ongoing Trial | RMS, PPMS | New gadolinium (Gd)-enhancing T1 lesion rate observed on magnetic resonance imaging (MRI) scans of the brain over 12 weeks | Yes | New or enlarging T2-weighted lesion rate observed on MRI scans of the brain over 12 weeks | Yes | NCT05119569 | |
| Proportion of participants free from any new Gd-enhancing T1 lesions and new or enlarging T2-weighted lesions observed on MRI scans of the brain over 12 weeks | Yes | |||||||||
| Number of participants with adverse events (AEs) and serious adverse events (SAEs) | Not yet—ongoing | |||||||||
| Number of participants with AEs and SAEs | Not yet—ongoing | |||||||||
| Number of participants with post-baseline suicidal ideation or suicidal behavior as measured using Columbia Suicide Severity Rating Scale (C-SSRS) | Not yet—ongoing | |||||||||
| Fenebrutinib | III | Roche–Genentech, Basel, Switzerland | Ongoing trial | Study 1: 746 Study 2: 751 | RMS compared to teriflunomide | Annualized relapse rate (ARR) | Not yet—ongoing | Time-to-onset of composite 12-week confirmed disability progression (cCDP12) | Not yet—ongoing | NCT04586010 NCT04586023 |
| Time-to-onset of composite 24-week confirmed disability progression (cCDP24) | Not yet—ongoing | |||||||||
| Time-to-onset of 12-week confirmed disability progression (CDP12) | Not yet—ongoing | |||||||||
| Time-to-onset of 24-week confirmed disability progression (CDP24) | Not yet—ongoing | |||||||||
| Total number of T1 gadolinium-enhancing (Gd+) lesions, new and/or enlarging T2-weighted lesions as detected by magnetic resonance imaging (MRI) | Not yet—ongoing | |||||||||
| Percentage change in total brain volume from week 24 as assessed by MRI | Not yet—ongoing | |||||||||
| Change in participant-reported physical impacts of multiple sclerosis (MS) measured by the Multiple Sclerosis, 29-Item [MSIS-29] Physical Scale | Not yet—ongoing | |||||||||
| Time-to-onset of 12-week confirmed 4-point worsening in Symbol Digit Modality Test (SDMT) Score | Not yet—ongoing | |||||||||
| Change from baseline to week 48 in the concentration of serum neurofilament light chain (NfL) | Not yet—ongoing | |||||||||
| Percentage of participants with adverse events (AEs) | Not yet—ongoing | |||||||||
| Plasma concentrations of fenebrutinib at specified timepoints | Not yet—ongoing | |||||||||
| Fenebrutinib | III | Roche–Genentech, Basel, Switzerland | Ongoing trial | 985 | PPMS compared to Ocrevus | Time-to-onset of composite 12-week confirmed disability progression (cCDP12) | Not yet—ongoing | Time-to-onset of composite 24-week CDP (cCDP24) | Not yet—ongoing | NCT04544449 |
| Time-to-onset of 12-week CDP (CDP12) | Not yet—ongoing | |||||||||
| Time-to-onset of 24-week CDP (CDP24) | Not yet—ongoing | |||||||||
| Percentage change in total brain volume assessed by magnetic resonance imaging (MRI) | Not yet—ongoing | |||||||||
| Change from baseline in participant-reported physical impacts of multiple sclerosis (MS) measured by the Multiple Sclerosis Impact Scale, 29-Item [MSIS-29] Physical Scale | Not yet—ongoing | |||||||||
| Time-to-onset of 12-week confirmed 4-point worsening in Symbol Digit Modality Test (SDMT) Score | Not yet—ongoing | |||||||||
| Percentage of participants with adverse events (AEs) | Not yet—ongoing | |||||||||
| Plasma concentrations of fenebrutinib at specified timepoints | Not yet—ongoing | |||||||||
| Percent change from screening in serum neurofilament light chain (NfL) levels | Not yet—ongoing | |||||||||
| Orelabrutinib | II | InnoCare/Biogen, Beijing, China | Ongoing trial | 160 | RRMS | The cumulative number of new GdE T1 MRI brain lesions | Yes | Incidence of treatment-emergent adverse events | Not yet—ongoing | NCT04711148 |
| Remibrutinib | III | Novartis, Basel, Switzerland | Ongoing trial | REMODEL:800 (estimated) | RMS | Annualized relapse rate (ARR) of confirmed relapses | Not Yet—ongoing | Time to 3-month confirmed disability progression (3mCDP) on Expanded Disability Status Scale (EDSS) | Not yet—ongoing | NCT05156281 |
| Time to 6-month confirmed disability progression (6mCDP) on EDSS | Not yet—ongoing | |||||||||
| Annualized rate of new or enlarging T2 lesion | Not yet—ongoing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reidy, M.; Khan, M.; Mills, E.A.; Wu, Q.; Garton, J.; Draayer, D.E.; Zahoor, I.; Giri, S.; Axtell, R.C.; Mao-Draayer, Y. New Frontiers in Multiple Sclerosis Treatment: From Targeting Costimulatory Molecules to Bispecific Antibodies. Int. J. Mol. Sci. 2025, 26, 3880. https://doi.org/10.3390/ijms26083880
Reidy M, Khan M, Mills EA, Wu Q, Garton J, Draayer DE, Zahoor I, Giri S, Axtell RC, Mao-Draayer Y. New Frontiers in Multiple Sclerosis Treatment: From Targeting Costimulatory Molecules to Bispecific Antibodies. International Journal of Molecular Sciences. 2025; 26(8):3880. https://doi.org/10.3390/ijms26083880
Chicago/Turabian StyleReidy, Megan, Meerah Khan, Elizabeth A. Mills, Qi Wu, Josh Garton, Dean E. Draayer, Insha Zahoor, Shailendra Giri, Robert C. Axtell, and Yang Mao-Draayer. 2025. "New Frontiers in Multiple Sclerosis Treatment: From Targeting Costimulatory Molecules to Bispecific Antibodies" International Journal of Molecular Sciences 26, no. 8: 3880. https://doi.org/10.3390/ijms26083880
APA StyleReidy, M., Khan, M., Mills, E. A., Wu, Q., Garton, J., Draayer, D. E., Zahoor, I., Giri, S., Axtell, R. C., & Mao-Draayer, Y. (2025). New Frontiers in Multiple Sclerosis Treatment: From Targeting Costimulatory Molecules to Bispecific Antibodies. International Journal of Molecular Sciences, 26(8), 3880. https://doi.org/10.3390/ijms26083880