
The Unified Theory of Neurodegeneration Pathogenesis Based on Axon Deamidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- 4E-BP2 plays a crucial role in memory formation, which is lost during neurodegeneration, as reported by Banko et al [7].
- 4E-BP2 function is disrupted by deamidation, as shown by Bidinosti et al. [8].
- 4E-BPs are involved in translational control, as discovered by Lin et al. [6].
- 4E-BP2 deamidation is implicated in immune responses to oxidative stress, as described by Reisz et al. [3].
- (1)
- Use my discovery [10] on the mechanism behind neuron-specific 4E-BP2 deamidation to link and bridge these four fields.
- (2)
- Develop a Unified Theory, which establishes that the regulation of axon-specific 4E-BP2 deamidation rates controls the occurrence and progression of neurodegenerative diseases.
2. Deamidation
- (1)
- Access to the solvent for main chain nitrogen deprotonation
- (2)
- Flexibility of the asparaginyl residue
- (1)
- Increased protein misfolding causes protein structure to break down, allowing for increased flexibility in peptide chains
- (2)
- Increased exposure to the solvent increases the susceptibility of main chain nitrogen deprotonation from hydroxyl groups found in the solvent.
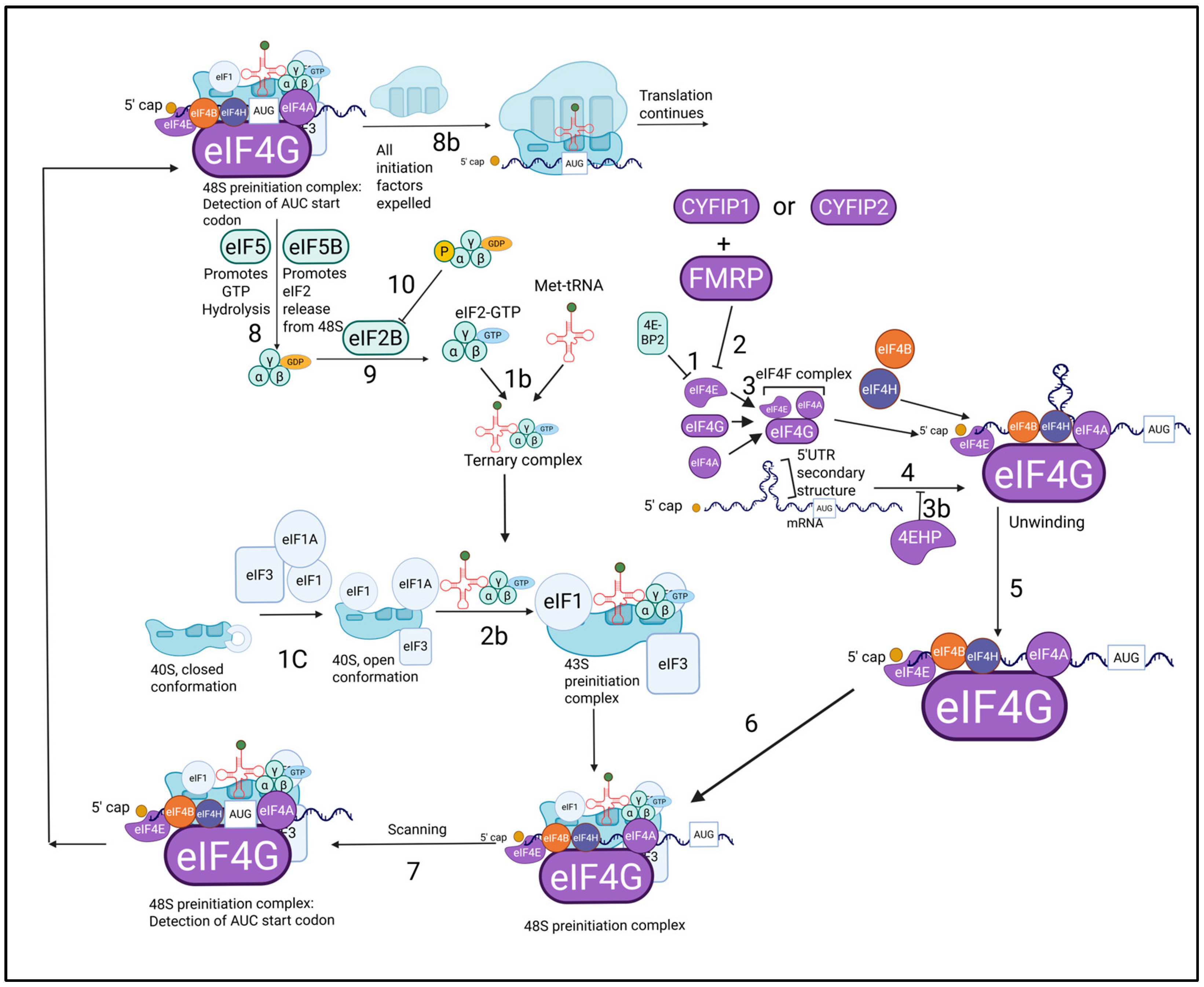
3. Translational Control
4. Neurodegeneration and Oxidative Stress
5. My Discovery
- (1)
- Kouloulia et al. [9] showed that proteasomal inhibition increases 4E-BP2 deamidation.
- (2)
- Sun et al. [189] reported that both axons and dendrites are proteasome-poor environments, through immunolabelling of rat cortical neurons and immunofluorescence staining of the mouse stratum pyramidale in the hippocampus.
- My experimental results found no evidence of deamidation rates increasing as axon length increased. On the contrary, the average deamidation ratio was higher in the shorter optic nerve axons (ratio = 0.7539) than in the longer sciatic nerve axons (0.4112).
- There is no long-term transport conducted within erythrocytes (red blood cells). However, as per Rolfs et al. [205], proteins specific to erythrocytes, such as hemoglobin, exhibit significantly longer half-lives, especially compared to proteins found in plasma. According to Smith et al. [206], plasma proteins undergo extracellular transport, yet I found no evidence that the transport itself significantly increases the transported proteins’ half-life.
- (1)
- The findings of Ritchie Truong [220] provide further evidence in support of my principle. Truong [220] concluded that 4E-BP2 deamidation was significant in skeletal muscle tissue but not in the heart muscle. As per Ruff et al. [221], skeletal muscle contraction depends on innervation via the axons found in neuromuscular junctions.
- (2)
- Based on Ruff et al. [221], my analysis shows that axon terminals are abundant in skeletal muscle tissue, which explains the presence of significant 4E-BP2 deamidation in muscle tissue. As described by His et al. [222] and Difrancesco et al. [223], cardiac muscle propulsion, however, is independent from motor axon input from the nervous system, with specialized myocardium cells being able to spontaneously generate action potentials.
- (1)
- Robinson et al. [2] described that deamidation leads to the programmed dysfunction of proteins
- (2)
- Bidinosti et al. [8] discovered that deamidation occurs in 4E-BP2, which functions as a translational inhibitor. He also reported that 4E-BP2 deamidation reduces the speed and efficiency of 4E-BP2 deamidation.
- (3)
- Tiwari et al. [88] demonstrated that translational control dysregulation from 4E-BP proteins like CYFIP2 causes the early development of neurodegenerative diseases such as Alzheimer’s.
- (4)
- Joseph [10] discovered that 4E-BP2 deamidation is caused by the proteasome-poor environment in neuronal projections, consisting mainly of axons.
- (5)
- (6)
- (7)
- Kouloulia et al. [9] found that deamidated 4E-BP2 overexpression is caused by proteasome inhibition and has been shown to decrease the expression of many genes implicated in cerebral cortex development and mitochondrial function, and that deamidated 4E-BP2 also decreases the activity of NF-κB in neurons.
- (8)
- Sorrentino et al. [125] asserted that mitochondrial dysfunction has been found to contribute to neuron cell death in Alzheimer’s and Parkinson’s
- (9)
- Guo et al. [224] explained that mitochondrial dysfunction causes oxidative stress.
- (10)
- Fang et al. [114] showed that axons are most vulnerable to oxidative stress
- (11)
- (12)
- Tain et al. [182] described that 4E-BPs inhibit PINK1 and Parkin protein mutants responsible for mitophagy disruption and dopaminergic neuron degradation in Alzheimer’s.
- (13)
- Kaltschmidt et al. [135] postulated that NF-κB protects neurons from necroptosis.
- (14)
- Meffert et al. [136] reported that NF-κB activation depends on synaptic transmission, and that NF-κB inactivation leads to learning and memory impairment.
- (15)
- Tiwari et al. [88], Li et al. [72] and Tong et al. [185] demonstrated that amyloid precursor protein and tau (both involved in Alzheimer’s pathology) as well as alpha-synuclein (involved in Parkinson’s pathology) all undergo cap-dependant protein translation involving eIF4E that is inhibited by 4E-BPs.
- (16)
- Creus-Muncunill et al. [187] reported that excessive protein synthesis after 4E-BP inhibition results in neurodegeneration in diseases such as Huntington’s disease.
- (1)
- Aβ aggregation and synapse loss in Alzheimer’s.
- (2)
- The formation of neurofibrillary tangles and resulting memory loss in Alzheimer’s
- (3)
- The formation of Lewy bodies in Parkinson’s
- (4)
- Dysfunctional mitochondria accumulation, which causes oxidative stress in Alzheimer’s and Parkinson’s
- (5)
- Early neurodegeneration in axons and synapses
- (6)
- A decrease in synaptic transmission induced NF-κB activity, which results in neuron death
6. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flatmark, T. On Heterogeneity of Beef Heart Cytochrome Ci Separation and Isolation of Subfractions by Disc Electrophoresis and Column Chromatography. Acta Chem. Scand. 1964, 18, 1656–1666. [Google Scholar] [CrossRef]
- Robinson, A.B.; McKerrow, J.H.; Legaz, M. Sequence dependent deamidation rates for model peptides of cytochrome C. Int. J. Pept. Protein Res. 1974, 6, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Reisz, J.A.; Nemkov, T.; Dzieciatkowska, M.; Culp-Hill, R.; Stefanoni, D.; Hill, R.C.; Yoshida, T.; Dunham, A.; Kanias, T.; Dumont, L.J.; et al. Methylation of protein aspartates and deamidated asparagines as a function of blood bank storage and oxidative stress in human red blood cells. Transfusion 2018, 58, 2978–2991. [Google Scholar] [CrossRef]
- Filipowicz, W.; Furuichi, Y.; Sierra, J.M.; Muthukrishnan, S.; Shatkin, A.J.; Ochoa, S. A protein binding the methylated 5′-terminal sequence, m7GpppN, of eukaryotic messenger RNA. Proc. Natl. Acad. Sci. USA 1976, 73, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Haghighat, A.; Sonenberg, N. eIF4G dramatically enhances the binding of eIF4E to the mRNA 5′-cap structure. J. Biol. Chem. 1997, 272, 21677–21680. [Google Scholar] [CrossRef]
- Lin, T.A.; Kong, X.; Haystead, T.A.; Pause, A.; Belsham, G.; Sonenberg, N.; Lawrence, J.C., Jr. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 1994, 266, 653–656. [Google Scholar] [CrossRef]
- Banko, J.L.; Poulin, F.; Hou, L.; DeMaria, C.T.; Sonenberg, N.; Klann, E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J. Neurosci. 2005, 25, 9581–9590. [Google Scholar] [CrossRef]
- Bidinosti, M.; Ran, I.; Sanchez-Carbente, M.R.; Martineau, Y.; Gingras, A.C.; Gkogkas, C.; Raught, B.; Bramham, C.R.; Sossin, W.S.; Costa-Mattioli, M.; et al. Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol. Cell 2010, 37, 797–808. [Google Scholar] [CrossRef]
- Kouloulia, S.; Hallin, E.I.; Simbriger, K.; Amorim, I.S.; Lach, G.; Amvrosiadis, T.; Chalkiadaki, K.; Kampaite, A.; Truong, V.T.; Hooshmandi, M.; et al. Raptor-mediated proteasomal degradation of deamidated 4E-BP2 regulates postnatal neuronal translation and NF-κB activity. Cell Rep. 2019, 29, 3620–3635. [Google Scholar] [CrossRef]
- Joseph, D. The Fundamental Neurobiological Mechanism of Oxidative Stress-Related 4E-BP2 Protein Deamidation. Int. J. Mol. Sci. 2024, 25, 12268. [Google Scholar] [CrossRef]
- Ow, Y.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Flatmark, T.; Sletten, K. Multiple forms of cytochrome c in the rat: Precursor-product relationship between the main component Cy I and the minor components Cy II and Cy III in vivo. J. Biol. Chem. 1968, 243, 1623–1629. [Google Scholar] [CrossRef]
- Lai, C.Y.; Chen, C.; Horecker, B.L. Primary structure of two COOH-terminal hexapeptides from rabbit muscle aldolase: A difference in the structure of the α and β subunits. Biochem. Biophys. Res. Commun. 1970, 40, 461–468. [Google Scholar] [CrossRef]
- Du, S.; Guan, Z.; Hao, L.; Song, Y.; Wang, L.; Gong, L.; Liu, L.; Qi, X.; Hou, Z.; Shao, S. Fructose-Bisphosphate Aldolase A Is a Potential Metastasis-Associated Marker of Lung Squamous Cell Carcinoma and Promotes Lung Cell Tumorigenesis and Migration. PLoS ONE 2014, 9, e85804. [Google Scholar] [CrossRef] [PubMed]
- Midelfort, C.F.; Mehler, A.H. Deamidation in vivo of an asparagine residue of rabbit muscle aldolase. Proc. Natl. Acad. Sci. USA 1972, 69, 1816–1819. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Robinson, A.B. Primary sequence dependence of the deamidation of rabbit muscle aldolase. Science 1974, 183, 85. [Google Scholar] [CrossRef]
- Morse, D.E.; Chan, W.; Horecker, B.L. The subunit structure and carboxy-terminal sequence of rabbit muscle aldolase. Proc. Natl. Acad. Sci. USA 1967, 58, 628–634. [Google Scholar] [CrossRef]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar]
- Robinson, A.B.; Scotchler, J.W. sequence dependent deamidation rates for model peptides of histone IV. Int. J. Pept. Protein Res. 1974, 6, 279–282. [Google Scholar] [CrossRef]
- Lindner, H.; Sarg, B.; Hoertnagl, B.; Helliger, W. The Microheterogeneity of the Mammalian H10Histone: EVIDENCE FOR AN AGE-DEPENDENT DEAMIDATION. J. Biol. Chem. 1998, 273, 13324–13330. [Google Scholar] [CrossRef]
- Medvedev, Z.A. Age changes of chromatin. A review. Mech. Ageing Dev. 1984, 28, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Solstad, T.; Flatmark, T. Microheterogeneity of recombinant human phenylalanine hydroxylase as a result of nonenzymatic deamidations of labile amide containing amino acids: Effects on catalytic and stability properties. Eur. J. Biochem. 2000, 267, 6302–6310. [Google Scholar] [CrossRef] [PubMed]
- Tomé, C.S.; Lopes, R.R.; Sousa, P.M.; Amaro, M.P.; Leandro, J.; Mertens, H.D.; Leandro, P.; Vicente, J.B. Structure of full-length wild-type human phenylalanine hydroxylase by small angle X-ray scattering reveals substrate-induced conformational stability. Sci. Rep. 2019, 9, 13615. [Google Scholar] [CrossRef]
- Takemoto, L.; Boyle, D. Specific glutamine and asparagine residues of γ-S crystallin are resistant to in vivo deamidation. J. Biol. Chem. 2000, 275, 26109–26112. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Kurimoto, E.; Oda, A. Mechanisms of deamidation of asparagine residues and effects of main-chain conformation on activation energy. Int. J. Mol. Sci. 2020, 21, 7035. [Google Scholar] [CrossRef]
- Yokoyama, H.; Mizutani, R.; Noguchi, S.; Hayashida, N. Structural and biochemical basis of the formation of isoaspartate in the complementarity-determining region of antibody 64M-5 Fab. Sci. Rep. 2019, 9, 18494. [Google Scholar] [CrossRef]
- Bidinosti, M. Identification and Characterisation of a Novel Posttranslational Modification of Translation Repressor 4E-BP2. Ph.D. Thesis, McGill University, Montreal, QC, Canada, 2009. [Google Scholar]
- Vlasak, J.; Bussat, M.C.; Wang, S.; Wagner-Rousset, E.; Schaefer, M.; Klinguer-Hamour, C.; Kirchmeier, M.; Corvaïa, M.; Ionescu, R.; Beck, A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal. Biochem. 2009, 392, 145–154. [Google Scholar] [CrossRef]
- Klein, D.R. Organic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2025. [Google Scholar]
- Takahashi, O.; Manabe, N.; Kirikoshi, R. A computational study of the mechanism of succinimide formation in the Asn–His sequence: Intramolecular catalysis by the His side chain. Molecules 2016, 21, 327. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Molecular clocks. Proc. Natl. Acad. Sci. USA 2001, 98, 944–949. [Google Scholar] [CrossRef]
- Capasso, S.; Salvadori, S. Effect of the three-dimensional structure on the deamidation reaction of ribonuclease A. J. Pept. Res. 1999, 54, 377–382. [Google Scholar] [CrossRef]
- Capasso, S.; Mazzarella, L.; Sica, F.; Zagari, A. Solid-state conformations of aminosuccinyl peptides: Crystal structure of tert-butyloxycarbonyl-l-leucyl-l-aminosuccinyl-l-phenylalaninamide. Biopolym. Orig. Res. Biomol. 1989, 28, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int. J. Pept. Protein Res. 1987, 30, 808–821. [Google Scholar] [CrossRef]
- Lapko, V.N.; Purkiss, A.G.; Smith, D.L.; Smith, J.B. Deamidation in human γS-crystallin from cataractous lenses is influenced by surface exposure. Biochemistry 2002, 41, 8638–8648. [Google Scholar] [CrossRef] [PubMed]
- Kirikoshi, R.; Manabe, N.; Takahashi, O. Succinimide formation from an NGR-containing cyclic peptide: Computational evidence for catalytic roles of phosphate buffer and the arginine side chain. Int. J. Mol. Sci. 2017, 18, 429. [Google Scholar] [CrossRef]
- Dunkelberger, E.B.; Buchanan, L.E.; Marek, P.; Cao, P.; Raleigh, D.P.; Zanni, M.T. Deamidation accelerates amyloid formation and alters amylin fiber structure. J. Am. Chem. Soc. 2012, 134, 12658–12667. [Google Scholar] [CrossRef]
- Slyvka, Y.I.; Goreshnik, E.A.; Ardan, B.R.; Veryasov, G.; Morozov, D.; Mys’kiv, M.G. A new tetranuclear copper (I) complex based on allyl (5-phenyl-1, 3, 4-thiadiazol-2-yl) azanide ligand: Synthesis and structural characterization. J. Mol. Struct. 2015, 1086, 125–130. [Google Scholar] [CrossRef]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [CrossRef]
- Böhme, L.; Bär, J.W.; Hoffmann, T.; Manhart, S.; Ludwig, H.H.; Rosche, F.; Demuth, H.U. Isoaspartate residues dramatically influence substrate recognition and turnover by proteases. Biol. Chem. 2008, 389, 1043–1053. [Google Scholar] [CrossRef]
- Murray, E.D.; Clarke, S. Synthetic peptide substrates for the erythrocyte protein carboxyl methyltransferase. Detection of a new site of methylation at isomerized L-aspartyl residues. J. Biol. Chem. 1984, 259, 10722–10732. [Google Scholar] [CrossRef]
- Emily, H.Y.C.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX-and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Cook, B.L.; Manson, S.R.; Niederhoff, R.A.; Langer, E.M.; Rosová, I.; Kulans, L.A.; Fu, X.; Weinberg, J.S.; Heinecke, J.W.; et al. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell 2002, 111, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.Q.; Yuksel, K.U.; Gracy, R.W. Terminal marking of triosephosphate isomerase: Consequences of deamidation. Arch. Biochem. Biophys. 1995, 322, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Matsuoka, Y.; Shirasawa, T. Biological significance of isoaspartate and its repair system. Biol. Pharm. Bull. 2005, 28, 1590–1596. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, M.L.; Schulze, S.E.; Lai, B.T.; Gray, H.B. Deamidation of α-synuclein. Protein Sci. 2009, 18, 1766–1773. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Sonenberg, N.; Morgan, M.A.; Merrick, W.C.; Shatkin, A.J. A polypeptide in eukaryotic initiation factors that crosslinks specifically to the 5′-terminal cap in mRNA. Proc. Natl. Acad. Sci. USA 1978, 75, 4843–4847. [Google Scholar] [CrossRef]
- Vanessa, M.A.J.O. Phosphorylation de la Protéine Liant les ARNs HuR/ELAVL1 par la Tyrosine Kinase Abelson: Implications sur la Fonction de HuR/ELAVL1 dans le Carcinome Hépatocellulaire. Ph.D. Thesis, Université Bordeaux, Bordeaux, France, 1983. [Google Scholar]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef]
- Lamphear, B.J.; Kirchweger, R.; Skern, T.; Rhoads, R.E. Mapping of Functional Domains in Eukaryotic Protein Synthesis Initiation Factor 4G (eIF4G) with Picornaviral Proteases: Implications for Cap-Dependent and Cap-Independent Translational Initiation (∗). J. Biol. Chem. 1995, 270, 21975–21983. [Google Scholar] [CrossRef]
- Ray, B.K.; Lawson, T.G.; Kramer, J.C.; Cladaras, M.H.; Grifo, J.A.; Abramson, R.D.; Merrick, W.C.; Thach, R.E. ATP-dependent unwinding of messenger RNA structure by eukaryotic initiation factors. J. Biol. Chem. 1985, 260, 7651–7658. [Google Scholar] [CrossRef]
- Sachs, A.B.; Sarnow, P.; Hentze, M.W. Starting at the beginning, middle, and end: Translation initiation in eukaryotes. Cell 1997, 89, 831–838. [Google Scholar] [CrossRef]
- Rogers, G.W.; Richter, N.J.; Lima, W.F.; Merrick, W.C. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J. Biol. Chem. 2001, 276, 30914–30922. [Google Scholar] [CrossRef] [PubMed]
- Cencic, R.; Im, Y.K.; Naineni, S.K.; Moustafa-Kamal, M.; Jovanovic, P.; Sabourin, V.; Annis, M.G.; Robert, F.; Scheing, T.M.; Koromilas, A.; et al. A second-generation eIF4A RNA helicase inhibitor exploits translational reprogramming as a vulnerability in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2024, 121, e2318093121. [Google Scholar] [CrossRef] [PubMed]
- Richter, N.J.; Rogers, G.W.; Hensold, J.O.; Merrick, W.C. Further biochemical and kinetic characterization of human eukaryotic initiation factor 4H. J. Biol. Chem. 1999, 274, 35415–35424. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Yan, R.; Rhoads, R.E. In vitro synthesis of human protein synthesis initiation factor 4 gamma and its localization on 43 and 48 S initiation complexes. J. Biol. Chem. 1994, 269, 2048–2055. [Google Scholar] [CrossRef]
- Lee, K.A.; Sonenberg, N. Inactivation of cap-binding proteins accompanies the shut-off of host protein synthesis by poliovirus. Proc. Natl. Acad. Sci. USA 1982, 79, 3447–3451. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. Photochemical cross-linking of cap binding proteins to eucaryotic mRNAs: Effect of mRNA 5′ secondary structure. Mol. Cell. Biol. 1985, 5, 3222–3230. [Google Scholar]
- Grifo, J.A.; Tahara, S.M.; Morgan, M.A.; Shatkin, A.J.; Merrick, W.C. New initiation factor activity required for globin mRNA translation. J. Biol. Chem. 1983, 258, 5804–5810. [Google Scholar] [CrossRef]
- Gao, M.; Rychlik, W.; Rhoads, R.E. Cloning and characterization of human eIF4E genes. J. Biol. Chem. 1998, 273, 4622–4628. [Google Scholar] [CrossRef]
- Mao, M.; Fu, G.; Wu, S.; Zhang, H.; Zhou, J.; Kan, X.; Huang, H.; He, L.; Gu, W.; Han, G.; et al. Identification of genes expressed in human CD34+ hematopoietic stem/progenitor cells by expressed sequence tags and efficient full-length cDNA cloning. Proc. Natl. Acad. Sci. USA 1998, 95, 8175. [Google Scholar] [CrossRef]
- Rom, E.; Kim, H.C.; Gingras, A.; Marcotrigiano, J.; Favre, D.; Olsen, H.; Burley, S.K.; Sonenberg, N. Cloning and Characterization of 4EHP, a Novel Mammalian eIF4E-related Cap-binding Protein. J. Biol. Chem. 1998, 273, 13104–13109. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Igreja, C. eIF4E-homologous protein (4EHP): A multifarious cap-binding protein. FEBS J. 2023, 290, 266–285. [Google Scholar] [CrossRef]
- Azpiazu, I.; Saltiel, A.R.; DePaoli-Roach, A.A.; John, C., Jr. Regulation of Both Glycogen Synthase and PHAS-I by Insulin in Rat Skeletal Muscle Involves Mitogen-activated Protein Kinase-independent and Rapamycin-sensitive Pathways (∗). J. Biol. Chem. 1996, 271, 5033–5039. [Google Scholar] [CrossRef]
- Mader, S.; Lee, H.; Pause, A.; Sonenberg, N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4γ and the translational repressors 4E-binding proteins. Mol. Cell. Biol. 1995, 15, 4990–4997. [Google Scholar] [CrossRef]
- Kimball, S.R.; Jurasinski, C.V.; Lawrence, J.C., Jr.; Jefferson, L.S. Insulin stimulates protein synthesis in skeletal muscle by enhancing the association of eIF-4E and eIF-4G. Am. J. Physiol.-Cell Physiol. 1997, 272, C754–C759. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, L.S.; Kimball, S.R.; Fadden, P.; Haystead, T.A.; Lawrence, J.C. Effects of insulin and diabetes on the association of eukaryotic initiation factor 4e and the translational regulator, phas-i, in rat skeletal muscle. FASEB J. 1996, 10, A739. [Google Scholar]
- Nusbaum, C.; Mikkelsen, T.S.; Zody, M.C.; Asakawa, S.; Taudien, S.; Garber, M.; Kodira, C.D.; Schueler, M.G.; Shimizu, A.; Whittaker, C.A.; et al. DNA sequence and analysis of human chromosome 8. Nature 2006, 439, 331–335. [Google Scholar] [CrossRef]
- Pugsley, L.; Naineni, S.K.; Amiri, M.; Yanagiya, A.; Cencic, R.; Sonenberg, N.; Pelletier, J. C8ORF88: A Novel eIF4E-Binding Protein. Genes 2023, 14, 2076. [Google Scholar] [CrossRef]
- Minich, W.B.; Balasta, M.L.; Goss, D.J.; Rhoads, R.E. Chromatographic resolution of in vivo phosphorylated and nonphosphorylated eukaryotic translation initiation factor eIF-4E: Increased cap affinity of the phosphorylated form. Proc. Natl. Acad. Sci. USA 1994, 91, 7668–7672. [Google Scholar] [CrossRef]
- Li, X.; An, W.L.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport 2004, 15, 2237–2240. [Google Scholar] [CrossRef]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richard, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 1993, 74, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Ascano, M.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Maurin, T.; Lebrigand, K.; Castagnola, S.; Paquet, A.; Jarjat, M.; Popa, A.; Grossi, M.; Rage, F.; Bardoni, B. HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res. 2018, 46, 6344–6355. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Ku, L.; Wilkinson, K.D.; Warren, S.T.; Feng, Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001, 29, 2276–2283. [Google Scholar] [CrossRef]
- Richter, J.D.; Bassell, G.J.; Klann, E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015, 16, 595–605. [Google Scholar] [CrossRef]
- Schenck, A.; Bardoni, B.; Moro, A.; Bagni, C.; Mandel, J.L. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. USA 2001, 98, 8844–8849. [Google Scholar] [CrossRef]
- Napoli, I.; Mercaldo, V.; Boyl, P.P.; Eleuteri, B.; Zalfa, F.; De Rubeis, S.; Di Marino, D.; Mohr, E.; Massimi, M.; Falconi, M.; et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 2008, 134, 1042–1054. [Google Scholar] [CrossRef]
- Clifton, N.E.; Thomas, K.L.; Wilkinson, L.S.; Hall, J.; Trent, S. FMRP and CYFIP1 at the synapse and their role in psychiatric vulnerability. Complex Psychiatry 2020, 6, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.C.; Szulc, B.R.; Drew, J.; Taylor, J.; Morgan, T.; Higgs, N.F.; López-Doménech, G.; Kittler, J.T. Autism and schizophrenia-associated CYFIP1 regulates the balance of synaptic excitation and inhibition. Cell Rep. 2019, 26, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Zhang, Y.; Kang, H.R.; Lee, S.H.; Shin, J.; Lee, C.H.; Kang, H.; Ma, R.; Jin, C.; Kim, Y.; et al. Altered presynaptic function and number of mitochondria in the medial prefrontal cortex of adult Cyfip2 heterozygous mice. Mol. Brain 2020, 13, 123. [Google Scholar] [CrossRef]
- Hsiao, K.; Harony-Nicolas, H.; Buxbaum, J.D.; Bozdagi-Gunal, O.; Benson, D.L. Cyfip1 regulates presynaptic activity during development. J. Neurosci. 2016, 36, 1564–1576. [Google Scholar] [CrossRef]
- Tiwari, S.S.; Mizuno, K.; Ghosh, A.; Aziz, W.; Troakes, C.; Daoud, J.; Golash, V.; Noble, W.; Hortobágyi, T.; Giese, K.P. Alzheimer-related decrease in CYFIP2 links amyloid production to tau hyperphosphorylation and memory loss. Brain 2016, 139, 2751–2765. [Google Scholar] [CrossRef]
- De Rubeis, S.; Pasciuto, E.; Li, K.W.; Fernández, E.; Di Marino, D.; Buzzi, A.; Ostroff, L.E.; Klann, E.; Zwartkruis, F.J.T.; Komiyama, N.H.; et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 2013, 79, 1169–1182. [Google Scholar] [CrossRef]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and control of the actin regulatory WAVE complex. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef] [PubMed]
- English, J.A.; Fan, Y.; Föcking, M.; Lopez, L.M.; Hryniewiecka, M.; Wynne, K.; Matigian, N.; Cagney, G.; Mackay-Sim, A.; Cotter, D.R. Reduced protein synthesis in schizophrenia patient-derived olfactory cells. Transl. Psychiatry 2015, 5, e663. [Google Scholar] [CrossRef]
- Kimball, S.R. Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 1999, 31, 25–29. [Google Scholar] [CrossRef]
- Pain, V.M. Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem. 1996, 236, 747–771. [Google Scholar] [CrossRef]
- Passmore, L.A.; Schmeing, T.M.; Maag, D.; Applefield, D.J.; Acker, M.G.; Algire, M.A.; Lorsch, J.R.; Ramakrishnan, V. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 2007, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Unbehaun, A.; Borukhov, S.I.; Hellen, C.U.; Pestova, T.V. Release of initiation factors from 48S complexes during ribosomal subunit joining and the link between establishment of codon-anticodon base-pairing and hydrolysis of eIF2-bound GTP. Genes Dev. 2004, 18, 3078–3093. [Google Scholar] [CrossRef] [PubMed]
- Pisarev, A.V.; Kolupaeva, V.G.; Pisareva, V.P.; Merrick, W.C.; Hellen, C.U.; Pestova, T.V. Specific functional interactions of nucleotides at key-3 and+ 4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 2006, 20, 624–636. [Google Scholar] [CrossRef]
- Pathania, M.; Davenport, E.C.; Muir, J.; Sheehan, D.F.; López-Doménech, G.; Kittler, J.T. The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl. Psychiatry 2014, 4, e374. [Google Scholar] [CrossRef]
- Haddon, J.E.; Titherage, D.; Heckenast, J.R.; Carter, J.; Owen, M.J.; Hall, J.; Wilkinson, L.W.; Jones, M.W. Linking haploinsufficiency of the autism-and schizophrenia-associated gene Cyfip1 with striatal-limbic-cortical network dysfunction and cognitive inflexibility. Transl. Psychiatry 2024, 14, 256. [Google Scholar] [CrossRef]
- Clifton, N.E.; Lin, J.Q.; Holt, C.E.; O’Donovan, M.C.; Mill, J. Enrichment of the local synaptic translatome for genetic risk associated with schizophrenia and autism spectrum disorder. Biol. Psychiatry 2024, 95, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef]
- Joffre, C.; Grégoire, S.; De Smedt, V.; Acar, N.; Bretillon, L.; Nadjar, A.; Layé, S. Modulation of brain PUFA content in different experimental models of mice. Prostaglandins Leukot. Essent. Fat. Acids 2016, 114, 1–10. [Google Scholar] [CrossRef]
- Carrié, I.; Clément, M.; de Javel, D.; Francès, H.; Bourre, J.M. Specific phospholipid fatty acid composition of brain regions in mice: Effects of n–3 polyunsaturated fatty acid deficiency and phospholipid supplementation. J. Lipid Res. 2000, 41, 465–472. [Google Scholar] [CrossRef]
- Chung, W.L.; Chen, J.J.; Su, H.M. Fish Oil Supplementation of Control and (n-3) Fatty Acid-Deficient Male Rats Enhances Reference and Working Memory Performance and Increases Brain Regional Docosahexaenoic Acid Levels3. J. Nutr. 2008, 138, 1165–1171. [Google Scholar] [CrossRef]
- Little, S.J.; Lynch, M.A.; Manku, M.; Nicolaou, A. Docosahexaenoic acid-induced changes in phospholipids in cortex of young and aged rats: A lipidomic analysis. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 155–162. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.K.; Carlson, S.E. Role of omega-3 fatty acids in brain development and function: Potential implications for the pathogenesis and prevention of psychopathology. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 329–349. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, Y.; Chen, Z.Y. Distribution, depletion and recovery of docosahexaenoic acid are region-specific in rat brain. Br. J. Nutr. 2005, 94, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L.; Close, D.R.; Stern, S.E. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 383–388. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Omar, R.A.; Kim, K.S.; Robakis, N.K. Immunohistochemical evidence of oxidative [corrected] stress in Alzheimer’s disease. Am. J. Pathol. 1992, 140, 621. [Google Scholar]
- Fang, C.; Bourdette, D.; Banker, G. Oxidative stress inhibits axonal transport: Implications for neurodegenerative diseases. Mol. Neurodegener. 2012, 7, 29. [Google Scholar] [CrossRef]
- Fusco, C.M.; Desch, K.; Dörrbaum, A.R.; Wang, M.; Staab, A.; Chan, I.C.; Vail, E.; Villeri, V.; Langer, J.D.; Schuman, E.M. Neuronal ribosomes exhibit dynamic and context-dependent exchange of ribosomal proteins. Nat. Commun. 2021, 12, 6127. [Google Scholar] [CrossRef]
- Keller, J.N.; Pang, Z.; Geddes, J.W.; Begley, J.G.; Germeyer, A.; Waeg, G.; Mattson, M.P. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 1997, 69, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, W.A.; Cashman, N.R.; Mattson, M.P. The lipid peroxidation product 4-hydroxynonenal impairs glutamate and glucose transport and choline acetyltransferase activity in NSC-19 motor neuron cells. Exp. Neurol. 1999, 155, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lodato, S.; Arlotta, P. Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell Dev. Biol. 2015, 31, 699–720. [Google Scholar] [CrossRef]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid β-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J. Neurosci. 1997, 17, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Strange, B.A.; Witter, M.P.; Lein, E.S.; Moser, E.I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 2014, 15, 655–669. [Google Scholar] [CrossRef]
- Mark, R.J.; Hensley, K.; Butterfield, D.A.; Mattson, M.P. Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 1995, 15, 6239–6249. [Google Scholar] [CrossRef]
- Poulsen, H.; Khandelia, H.; Morth, J.P.; Bublitz, M.; Mouritsen, O.G.; Egebjerg, J.; Nissen, P. Neurological disease mutations compromise a C-terminal ion pathway in the Na+/K+-ATPase. Nature 2010, 467, 99–102. [Google Scholar] [CrossRef]
- Kinoshita, P.F.; Marques Orellana, A.M.; Nakao, V.W.; Menezes Quintas, L.E.; Kawamoto, E.M.; Scavone, C. The Janus face of ouabain in Na+/K+-ATPase and calcium signalling in neurons. Br. J. Pharmacol. 2022, 179, 1512–1524. [Google Scholar] [CrossRef]
- Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Hudry, E.; Bacskai, B.J. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; Counts, S.E.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Mahdi, M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Jiao, Q.; Wei, M.; Wang, Y.; Cui, A.; Li, Z.; Li, G. Axonal mitophagy in retinal ganglion cells. Cell Commun. Signal. 2024, 22, 382. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife 2020, 9, e50260. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Czaniera, N.J.; Schulten, W.; Kaltschmidt, C. NF-κB in Alzheimer’s Disease: Friend or Foe? Opposite Functions in Neurons and Glial Cells. Int. J. Mol. Sci. 2024, 25, 11353. [Google Scholar] [CrossRef] [PubMed]
- Meffert, M.K.; Chang, J.M.; Wiltgen, B.J.; Fanselow, M.S.; Baltimore, D. NF-κB functions in synaptic signaling and behavior. Nat. Neurosci. 2003, 6, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ochoa, E.O.; Contreras, M.; Cseresnyés, Z.; Schneider, M.F. Ca2+ signal summation and NFATc1 nuclear translocation in sympathetic ganglion neurons during repetitive action potentials. Cell Calcium 2007, 41, 559–571. [Google Scholar] [CrossRef]
- Tamagno, E.; Robino, G.; Obbili, A.; Bardini, P.; Aragno, M.; Parola, M.; Danni, O. H2O2 and 4-hydroxynonenal mediate amyloid β-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp. Neurol. 2003, 180, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Donovan, N.; Becker, E.B.; Konishi, Y.; Bonni, A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 2002, 277, 40944–40949. [Google Scholar] [CrossRef]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK-and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef]
- Inoshita, S.; Takeda, K.; Hatai, T.; Terada, Y.; Sano, M.; Hata, J.; Umezaawa, A.; Ichijo, H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J. Biol. Chem. 2002, 277, 43730–43734. [Google Scholar] [CrossRef]
- Farley, N.; Pedraza-Alva, G.; Serrano-Gomez, D.; Nagaleekar, V.; Aronshtam, A.; Krahl, T.; Thornton, T.; Rincón, M. p38 mitogen-activated protein kinase mediates the Fas-induced mitochondrial death pathway in CD8+ T cells. Mol. Cell. Biol. 2006, 26, 2118–2129. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Long, J.; Li, W.; Wang, X.; Hu, N.; Zhao, X.; Sun, T. White matter changes in Parkinson’s disease. NPJ Park. Dis. 2023, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–949. [Google Scholar] [CrossRef]
- Ross, G.W.; Petrovitch, H.; Abbott, R.D.; Nelson, J.; Markesbery, W.; Davis, D.; Hardman, J.; Launer, L.; Masaki, K.; Tanner, C.M.; et al. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 56, 532–539. [Google Scholar] [CrossRef]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Bell, K.F.; Cuello, A.C. Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 2006, 545, 11–21. [Google Scholar] [CrossRef]
- Gilley, J.; Adalbert, R.; Coleman, M.P. Modelling early responses to neurodegenerative mutations in mice. Biochem. Soc. Trans. 2011, 39, 933–938. [Google Scholar] [CrossRef]
- Adalbert, R.; Nogradi, A.; Babetto, E.; Janeckova, L.; Walker, S.A.; Kerschensteiner, M.; Misgeld, T.; Coleman, M.P. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 2009, 132, 402–416. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Epis, R.; Di Luca, M. Amyloid flirting with synaptic failure: Towards a comprehensive view of Alzheimer’s disease pathogenesis. Eur. J. Pharmacol. 2008, 585, 109–118. [Google Scholar] [CrossRef]
- Westmark, C.J. What’s hAPPening at synapses? The role of amyloid β-protein precursor and β-amyloid in neurological disorders. Mol. Psychiatry 2013, 18, 425–434. [Google Scholar] [CrossRef]
- Tissot, C.; Therriault, J.; Pascoal, T.A.; Chamoun, M.; Lussier, F.Z.; Savard, M.; Mathotaarachchi, S.S.; Benedet, A.L.; Thomas, E.M.; Parsons, M.; et al. Association between regional tau pathology and neuropsychiatric symptoms in aging and dementia due to Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12154. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.J.; Song, C.; Deek, J.; Miller, H.P.; Li, Y.; Choi, M.C.; Wilson, L.; Feinstein, S.C.; Safinya, C.R. Tau mediates microtubule bundle architectures mimicking fascicles of microtubules found in the axon initial segment. Nat. Commun. 2016, 7, 12278. [Google Scholar] [CrossRef] [PubMed]
- Aronov, S.; Aranda, G.; Behar, L.; Ginzburg, I. Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J. Neurosci. 2001, 21, 6577–6587. [Google Scholar] [CrossRef]
- Aronov, S.; Aranda, G.; Behar, L.; Ginzburg, I. Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J. Cell Sci. 2002, 115, 3817–3827. [Google Scholar] [CrossRef]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic degeneration in Alzheimer disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar]
- KoSIK, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P.; Smith, C.; Couck, A.M.; Gallo, J.M.; Anderton, B.H. Developmental Changes in τ Phosphorylation: Fetal τ Is Transiently Phosphorylated in a Manner Similar to Paired Helical Filament-τ Characteristic of Alzheimer’s Disease. J. Neurochem. 1993, 61, 2071–2080. [Google Scholar] [CrossRef]
- Kenessey, A.; Yen, S.H.C. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993, 629, 40–46. [Google Scholar] [CrossRef]
- Yu, Y.; Run, X.; Liang, Z.; Li, Y.; Liu, F.; Liu, Y.; Izbal, K.; Grundke-Iqbal, I.; Gong, C.X. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 2009, 108, 1480–1494. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Kimura, T.; Ishiguro, K.; Hisanaga, S.I. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Jicha, G.A.; Weaver, C.; Lane, E.; Vianna, C.; Kress, Y.; Rockwood, J.; Davies, P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J. Neurosci. 1999, 19, 7486–7494. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Shi, J.; Yin, D.; El-Akkad, E.; Grundke-Iqbal, I.; Izbal, K.; Gong, C.X. PKA modulates GSK-3β-and cdk5-catalyzed phosphorylation of tau in site-and kinase-specific manners. FEBS Lett. 2006, 580, 6269–6274. [Google Scholar] [CrossRef]
- Ando, K.; Maruko-Otake, A.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Aβ42-induced tau toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef]
- Pellarin, I.; Dall’Acqua, A.; Favero, A.; Segatto, I.; Rossi, V.; Crestan, N.; Karimbayli, J.; Belletti, B.; Baldassarre, G. Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Signal Transduct. Target. Ther. 2025, 10, 11. [Google Scholar]
- Alonso, A.D.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Biernat, J.; Von Bergen, M.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Jury-Garfe, N.; Patel, H.; You, Y.; Perkins, A.; You, Y.; Lee-Gosselin, A.; Vidal, R.; Lasagna-Reeves, C.A. Phosphorylation at serine 214 correlates with tau seeding activity in an age-dependent manner in two mouse models for tauopathies and is required for tau transsynaptic propagation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Tran, A.; Turner, E.C.; Belfiore, R.; Branca, C.; Oddo, S. Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell 2018, 17, e12775. [Google Scholar] [CrossRef]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 2019, 29, 3592–3604. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; Deture, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Stephens, A.D.; Villegas, A.F.; Chung, C.W.; Vanderpoorten, O.; Pinotsi, D.; Mela, I.; Ward, E.; McCow, T.M.; Cubitt, R.; Routh, A.F.; et al. α-Synuclein fibril and synaptic vesicle interactions lead to vesicle destruction and increased lipid-associated fibril uptake into iPSC-derived neurons. Commun. Biol. 2023, 6, 526. [Google Scholar] [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The role of α-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Spano, M.; Signorelli, M.; Vitaliani, R.; Aguglia, E.; Giometto, B. The possible involvement of mitochondrial dysfunctions in Lewy body dementia: A systematic review. Funct. Neurol. 2015, 30, 151. [Google Scholar] [CrossRef]
- Tain, L.S.; Chowdhury, R.B.; Tao, R.N.; Moisoi, N.; Martins, L.M.; Downward, J.; Whitworth, A.J.; Tapon, N. Drosophila HtrA2 is dispensable for apoptosis but acts downstream of PINK1 independently from Parkin. Cell Death Differ. 2009, 16, 1118–1125. [Google Scholar] [CrossRef]
- Hoshino, T.; Mukai, A.; Yamashita, H.; Misawa, H.; Urushitani, M.; Tashiro, Y.; Urushitani, M.; Tashiro, Y.; Matsuzawa, S.; Takahashi, R. NDRG1 upregulation by ubiquitin proteasome system dysfunction aggravates neurodegeneration. Mol. Brain 2024, 17, 77. [Google Scholar] [CrossRef]
- Zhang, P.; Park, H.J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhang, P.; Yang, X.; Liu, X.; Zhang, J.; Grudniewska, M.; Jung, I.; Abegg, D.; Liu, J.; Childs-Disney, J.L.; et al. Decreasing the intrinsically disordered protein α-synuclein levels by targeting its structured mRNA with a ribonuclease-targeting chimera. Proc. Natl. Acad. Sci. USA 2024, 121, e2306682120. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Mishra, S.K.; Pant, H.C. Oxidative stress in neurodegeneration. Adv. Pharmacol. Pharm. Sci. USA 2011, 2011, 572634. [Google Scholar] [CrossRef] [PubMed]
- Creus-Muncunill, J.; Badillos-Rodríguez, R.; Garcia-Forn, M.; Masana, M.; Garcia-Díaz Barriga, G.; Guisado-Corcoll, A.; Alberch, J.; Malagelada, C.; Delgado-García, J.M.; Gruart, A.; et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 2019, 142, 3158–3175. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Yu, X.; Shelbourne, P.; Li, J. Huntingtin Aggregate-Associated Axonal Degeneration is an Early Pathological Event in Huntington’s Disease Mice. J. Neurosci. 2001, 21, 8473. [Google Scholar] [CrossRef]
- Sun, C.; Desch, K.; Nassim-Assir, B.; Giandomenico, S.L.; Nemcova, P.; Langer, J.D.; Schuman, E.M. An abundance of free regulatory (19 S) proteasome particles regulates neuronal synapses. Science 2023, 380, eadf2018. [Google Scholar] [CrossRef]
- Szabó, G.G.; Holderith, N.; Gulyás, A.I.; Freund, T.F.; Hájos, N. Distinct synaptic properties of perisomatic inhibitory cell types and their different modulation by cholinergic receptor activation in the CA3 region of the mouse hippocampus. Eur. J. Neurosci. 2010, 31, 2234–2246. [Google Scholar] [CrossRef]
- Ellender, T.J.; Paulsen, O. The many tunes of perisomatic targeting interneurons in the hippocampal network. Front. Cell. Neurosci. 2010, 4, 26. [Google Scholar] [CrossRef]
- Tyzio, R.; Represa, A.; Jorquera, I.; Ben-Ari, Y.; Gozlan, H.; Aniksztejn, L. The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 1999, 19, 10372–10382. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain—From eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef]
- Zhao, B.; Li, Y.; Fan, Z.; Wu, Z.; Shu, J.; Yang, X.; Yang, Y.; Wang, X.; Li, B.; Wang, X.; et al. Eye-brain connections revealed by multimodal retinal and brain imaging genetics. Nat. Commun. 2024, 15, 6064. [Google Scholar] [CrossRef]
- Butt, A.M.; Pugh, M.; Hubbard, P.; James, G. Functions of optic nerve glia: Axoglial signalling in physiology and pathology. Eye 2004, 18, 1110–1121. [Google Scholar] [CrossRef]
- Prakash, B.A.; Bhardwaj, A.K.; Devi, M.N.; Sridevi, N.S.; Rao, P.K.; Singh, G. Sciatic nerve division: A cadaver study in the Indian population and review of the literature. Singap. Med. J. 2010, 51, 721. [Google Scholar]
- Murakami, T.; Iwanaga, T.; Ogawa, Y.; Fujita, Y.; Sato, E.; Yoshitomi, H.; Sunada, Y.; Nakamura, A. Development of sensory neuropathy in streptozotocin-induced diabetic mice. Brain Behav. 2013, 3, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Priller, J.; Miron, V.E. Microglia regulate central nervous system myelin growth and integrity. Nature 2023, 613, 121. [Google Scholar]
- Schmalbruch, H. Fiber composition of the rat sciatic nerve. Anat. Rec. 1986, 215, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.K.; Wilcox, H.M.; Scott, R.W.; Siman, R. Proteasome inhibition enhances the stability of mouse CuZn superoxide dismutase with mutations linked to familial amyotrophic lateral sclerosis. J. Neurol. Sci. 1996, 139, 15–20. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Wong, P.C.; Becher, M.W.; Pardo, C.A.; Lee, M.K.; Xu, Z.S.; Thinakaran, G.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; et al. Axonal transport of mutant superoxide dismutase 1 and focal axonal abnormalities in the proximal axons of transgenic mice. Neurobiol. Dis. 1998, 5, 27–35. [Google Scholar] [CrossRef]
- Black, M.M.; Lasek, R.J. Axonal transport of actin: Slow component b is the principal source of actin for the axon. Brain Res. 1979, 171, 401–413. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Shi, Y.; Rhodes, N.R.; Abdolvahabi, A.; Kohn, T.; Cook, N.P.; Marti, A.A.; Shaw, B.F. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J. Am. Chem. Soc. 2013, 135, 15897–15908. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, Z.; Frey, B.L.; Shi, X.; Kawai, Y.; Smith, L.M.; Welham, N.V. An atlas of protein turnover rates in mouse tissues. Nat. Commun. 2021, 12, 6778. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.B. Transport, interactions and retention of plasma proteins in the intima: The barrier function of the internal elastic lamina. Eur. Heart J. 1990, 11 (Suppl. SE), 72–81. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Deamidation of human proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 12409–12413. [Google Scholar] [CrossRef]
- Xiao, N.; Xu, S.; Li, Z.; Tang, M.; Mao, R.; Yang, T.; Ma, S.; Wang, P.; Li, M.; Sunilkumar, A.; et al. A single photoreceptor splits perception and entrainment by cotransmission. Nature 2023, 623, 562–570. [Google Scholar] [CrossRef]
- Krames, E.S. The role of the dorsal root ganglion in the development of neuropathic pain. Pain Med. 2014, 15, 1669–1685. [Google Scholar] [CrossRef]
- Neelam, S.; Kakhniashvili, D.G.; Wilkens, S.; Levene, S.D.; Goodman, S.R. Functional 20S proteasomes in mature human red blood cells. Exp. Biol. Med. 2011, 236, 580–591. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Osmulski, P.A.; Gaczynska, M.; Glickman, M.H. The central unit within the 19S regulatory particle of the proteasome. Nat. Struct. Mol. Biol. 2008, 15, 573. [Google Scholar] [CrossRef]
- Sae-Lee, W.; McCafferty, C.L.; Verbeke, E.J.; Havugimana, P.C.; Papoulas, O.; McWhite, C.D.; Houser, J.R.; Vanuytsel, K.; Murphy, G.J.; Drew, K.; et al. The protein organization of a red blood cell. Cell Rep. 2022, 40, 111103. [Google Scholar] [CrossRef]
- Sahu, I.; Mali, S.M.; Sulkshane, P.; Xu, C.; Rozenberg, A.; Morag, R.; Sahoo, M.P.; Singh, S.K.; Ding, Z.; Wang, Y.; et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021, 12, 6173. [Google Scholar] [CrossRef]
- Minagar, A.; Ma, W.; Zhang, X.; Wang, X.; Zhang, K.; Alexander, J.S.; Gonzalez-Toldeo, E.; Albitar, M. Plasma ubiquitin–proteasome system profile in patients with multiple sclerosis: Correlation with clinical features, neuroimaging, and treatment with interferon-beta-1b. Neurol. Res. 2012, 34, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C. The mechanism of action of interferon-β in relapsing multiple sclerosis. CNS Drugs 2011, 25, 491–502. [Google Scholar] [CrossRef]
- Woo, M.S.; Engler, J.B.; Friese, M.A. The neuropathobiology of multiple sclerosis. Nat. Rev. Neurosci. 2024, 25, 493–513. [Google Scholar] [CrossRef] [PubMed]
- Neishabouri, A.M.; Faisal, A.A. The metabolic efficiency of myelinated vs unmyelinated axons. BMC Neurosci. 2011, 12 (Suppl. S1), P100. [Google Scholar] [CrossRef]
- Deng, Y.; Beahm, D.R.; Ionov, S.; Sarpeshkar, R. Measuring and modeling energy and power consumption in living microbial cells with a synthetic ATP reporter. BMC Biol. 2021, 19, 101. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440. [Google Scholar] [CrossRef]
- Truong, T.V. Indentifying the Mechanism Underlying Tissue Specific Deamidation of Translation Repressor 4E-BP2. Master’s Thesis, McGill University, Montreal, QC, Canada, 2017. [Google Scholar]
- Ruff, R.L. Neurophysiology of the neuromuscular junction: Overview. Ann. N. Y. Acad. Sci. 2003, 998, 1–10. [Google Scholar] [CrossRef]
- His, W. The activity of the embryonic human heart and its significance for the understanding of the heart movement in the adult. J. Hist. Med. Allied Sci. 1949, 4, 289–318. [Google Scholar] [CrossRef]
- DiFrancesco, D. The onset and autonomic regulation of cardiac pacemaker activity: Relevance of the f current. Cardiovasc. Res. 1995, 29, 449–456. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, D. The Unified Theory of Neurodegeneration Pathogenesis Based on Axon Deamidation. Int. J. Mol. Sci. 2025, 26, 4143. https://doi.org/10.3390/ijms26094143
Joseph D. The Unified Theory of Neurodegeneration Pathogenesis Based on Axon Deamidation. International Journal of Molecular Sciences. 2025; 26(9):4143. https://doi.org/10.3390/ijms26094143
Chicago/Turabian StyleJoseph, Davis. 2025. "The Unified Theory of Neurodegeneration Pathogenesis Based on Axon Deamidation" International Journal of Molecular Sciences 26, no. 9: 4143. https://doi.org/10.3390/ijms26094143
APA StyleJoseph, D. (2025). The Unified Theory of Neurodegeneration Pathogenesis Based on Axon Deamidation. International Journal of Molecular Sciences, 26(9), 4143. https://doi.org/10.3390/ijms26094143