Chemical Reactivity as Described by Quantum Chemical Methods

Abstract
:1. Quantum Mechanics, Quantum Chemistry, Computational Chemistry, Density Functional Theory: who is who
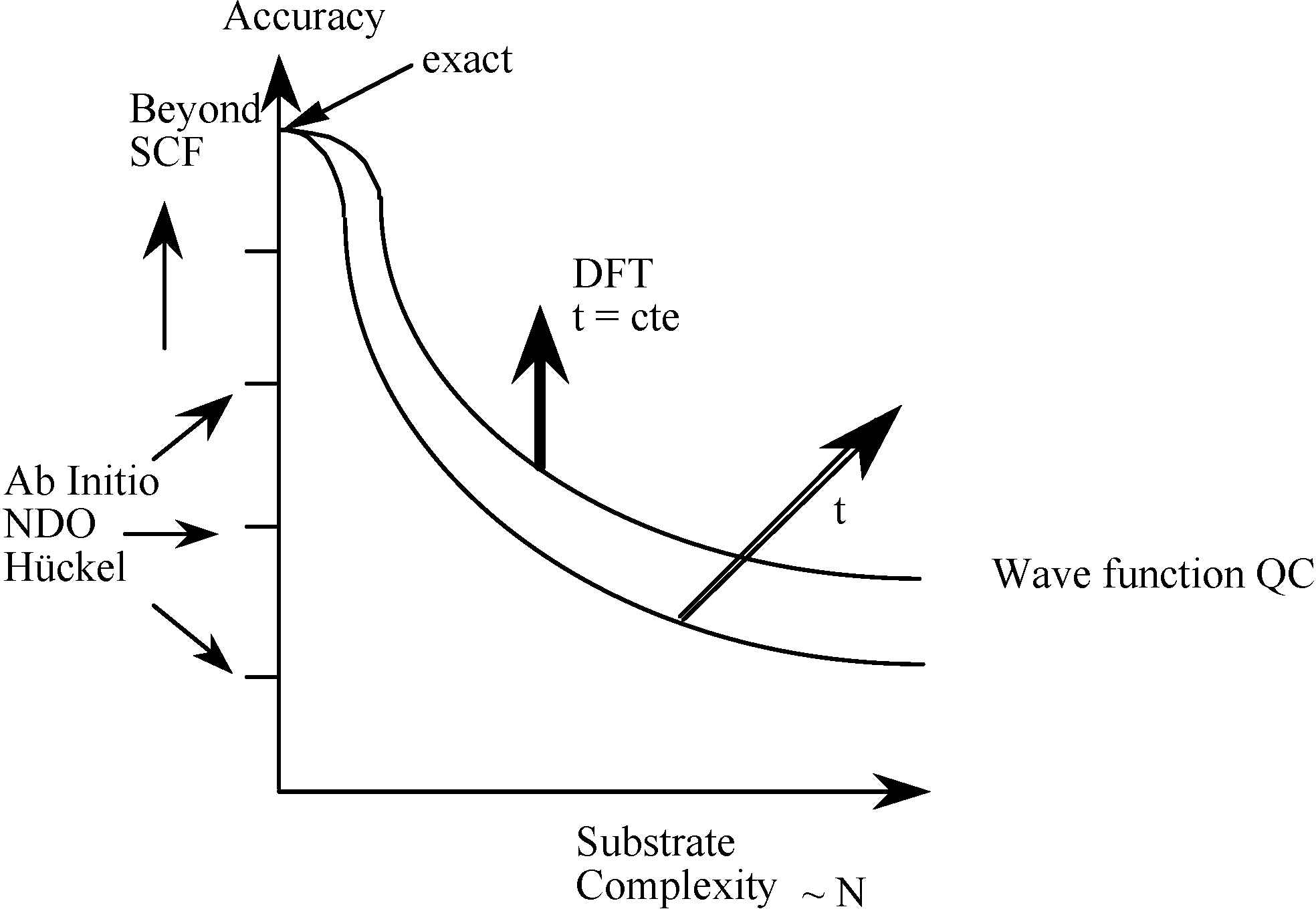
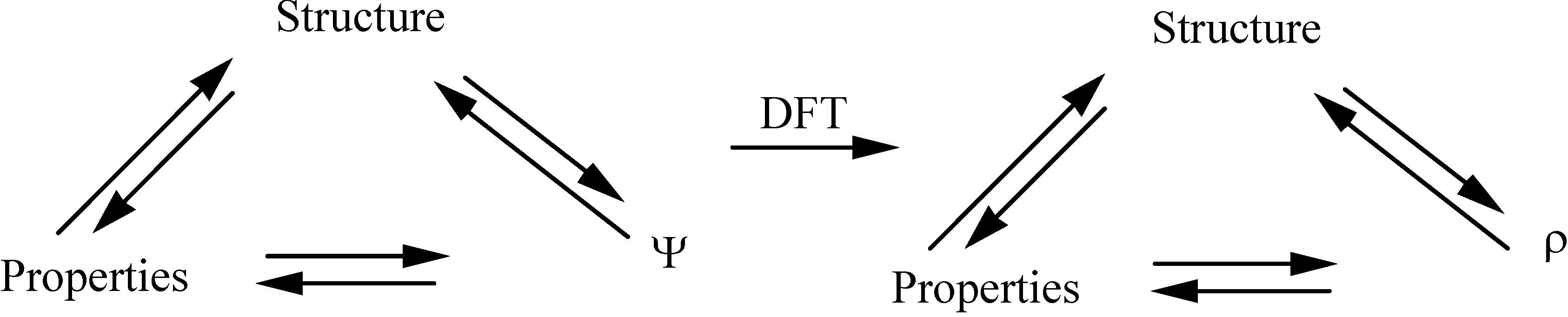
1.1. From Quantum Mechanics to Quantum Chemistry and Computational Chemistry
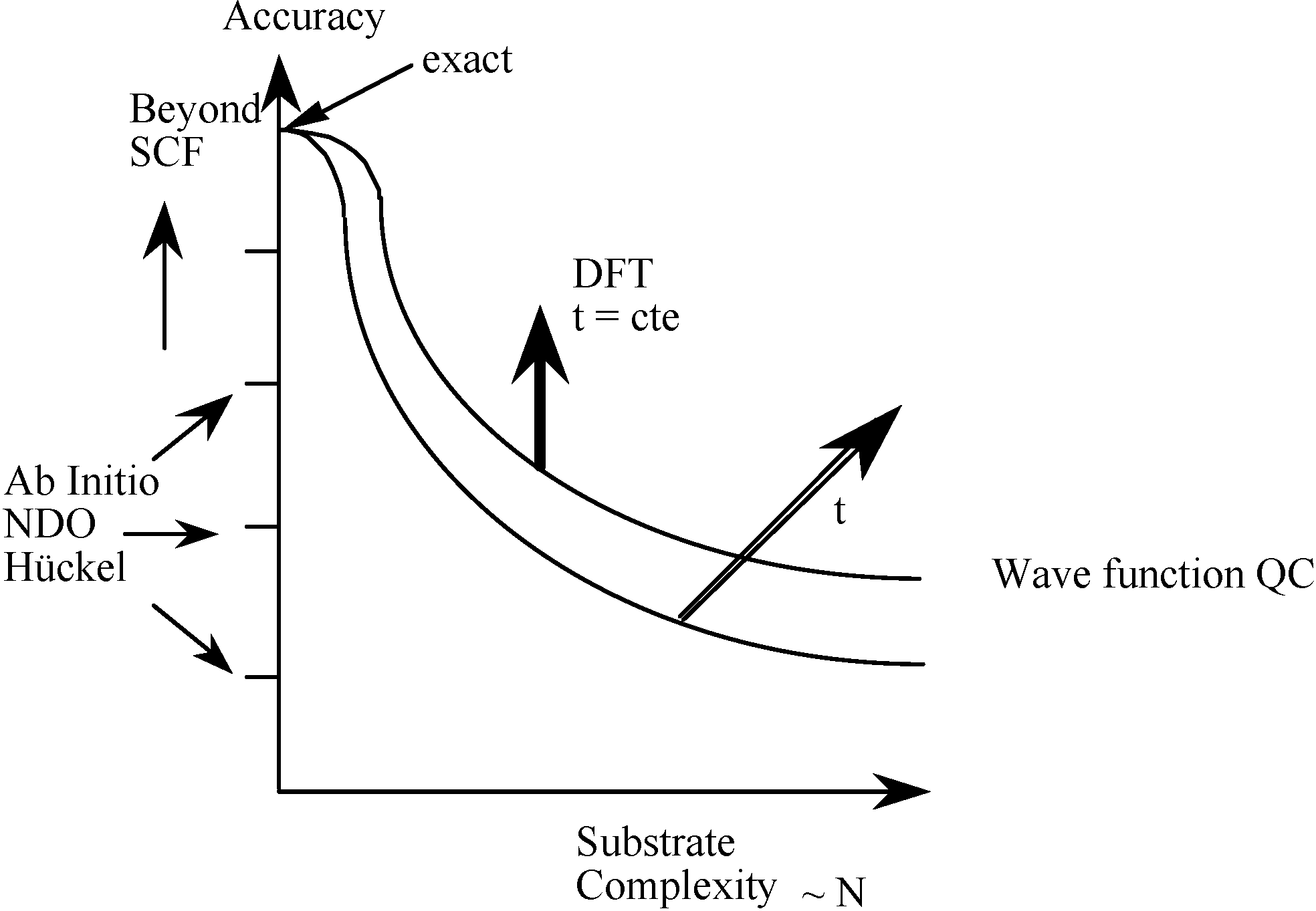
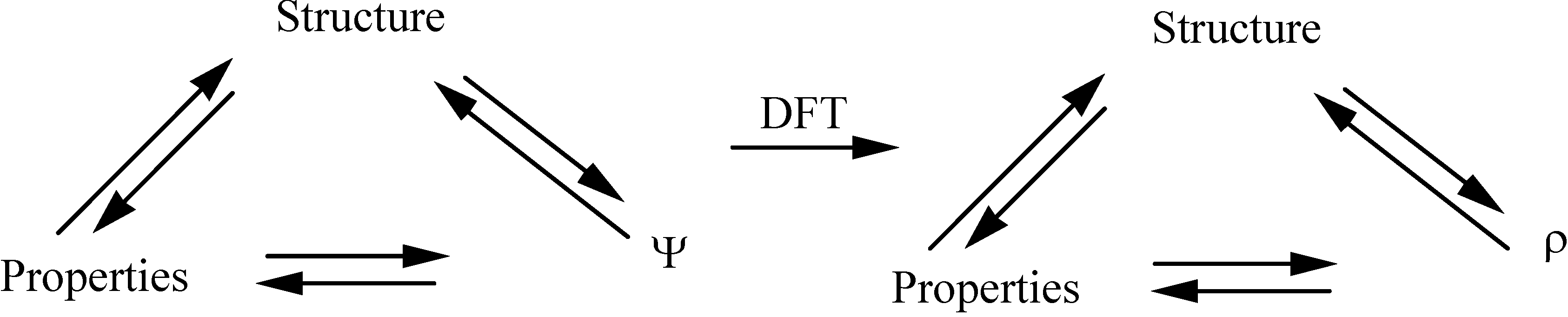
1.2. Density Functional Theory revolutionarized Quantum Chemistry from a computational point of view
1.3. DFT as a provider of new insights : conceptual DFT
1.3.1. From computational chemistry to chemical insight
1.3.2. DFT as a source of Chemical Concepts

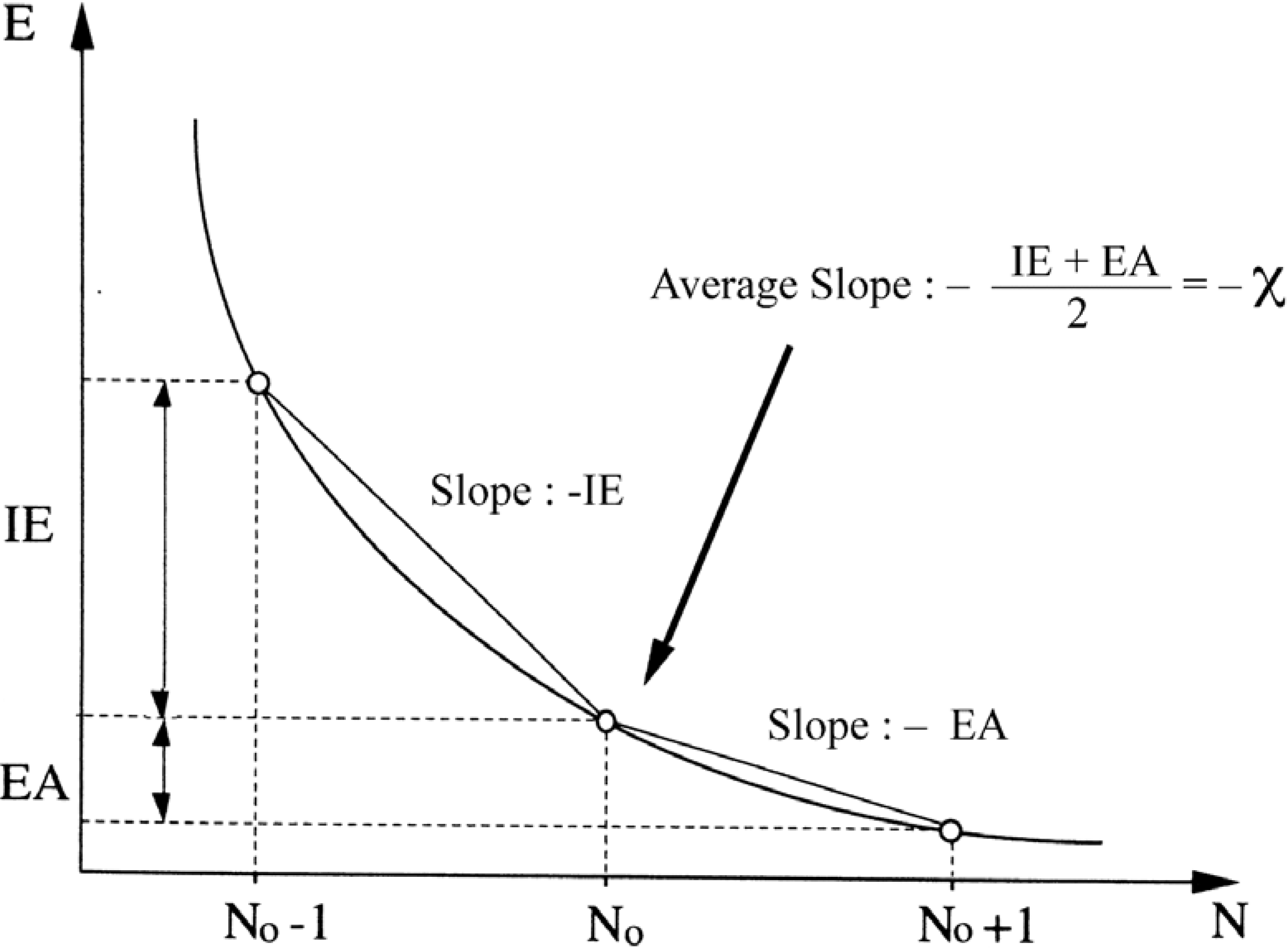
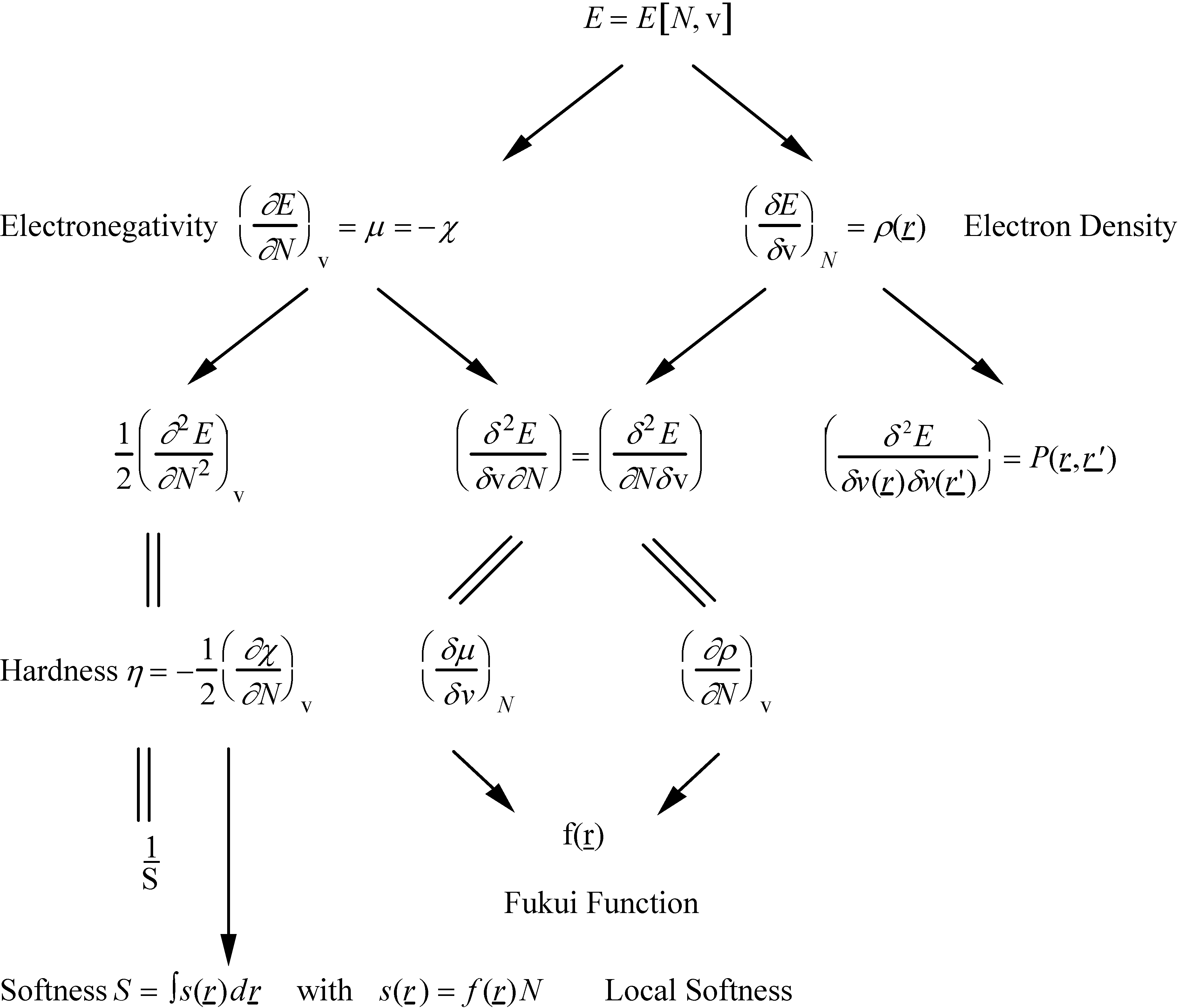
- -
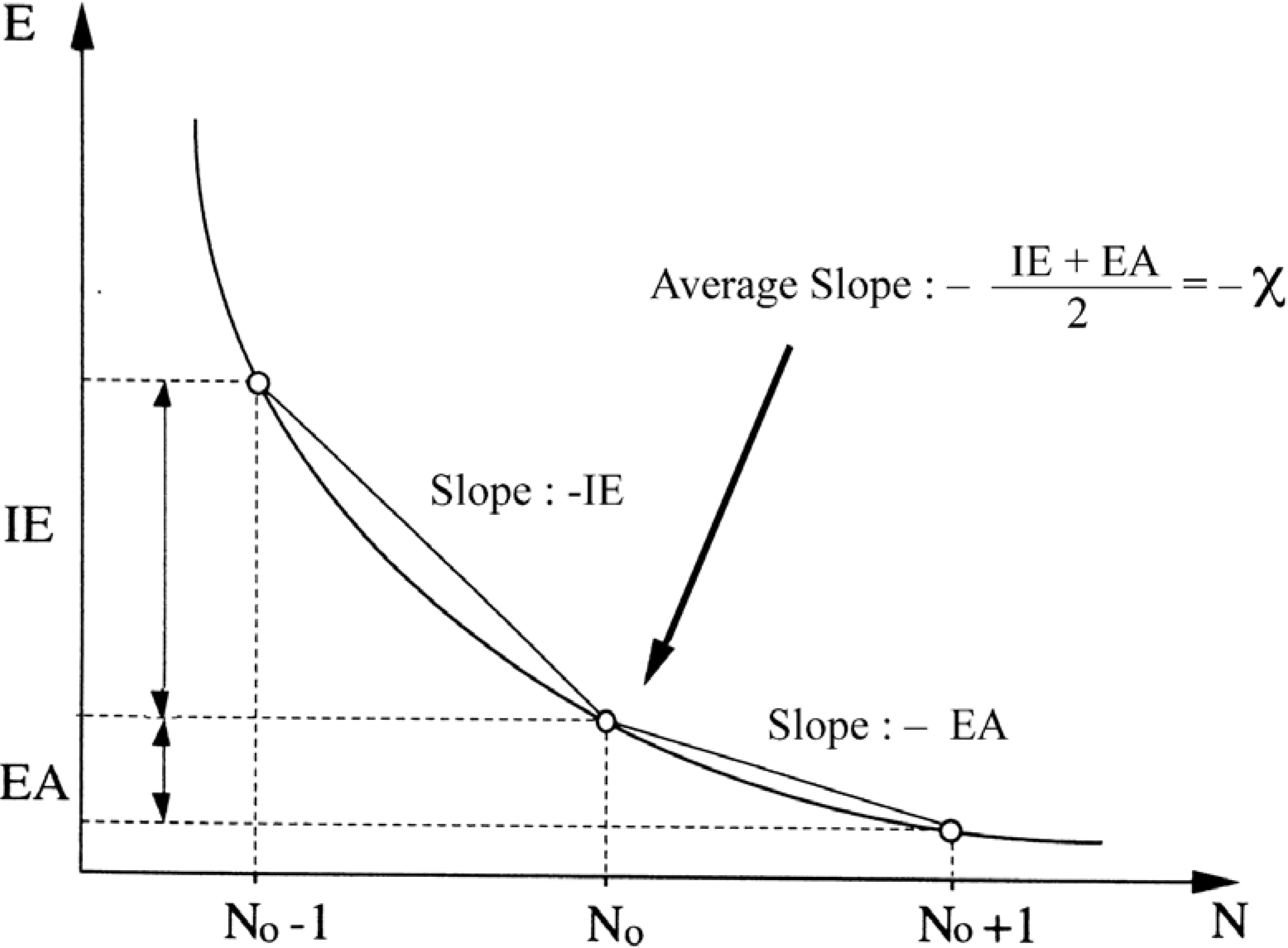
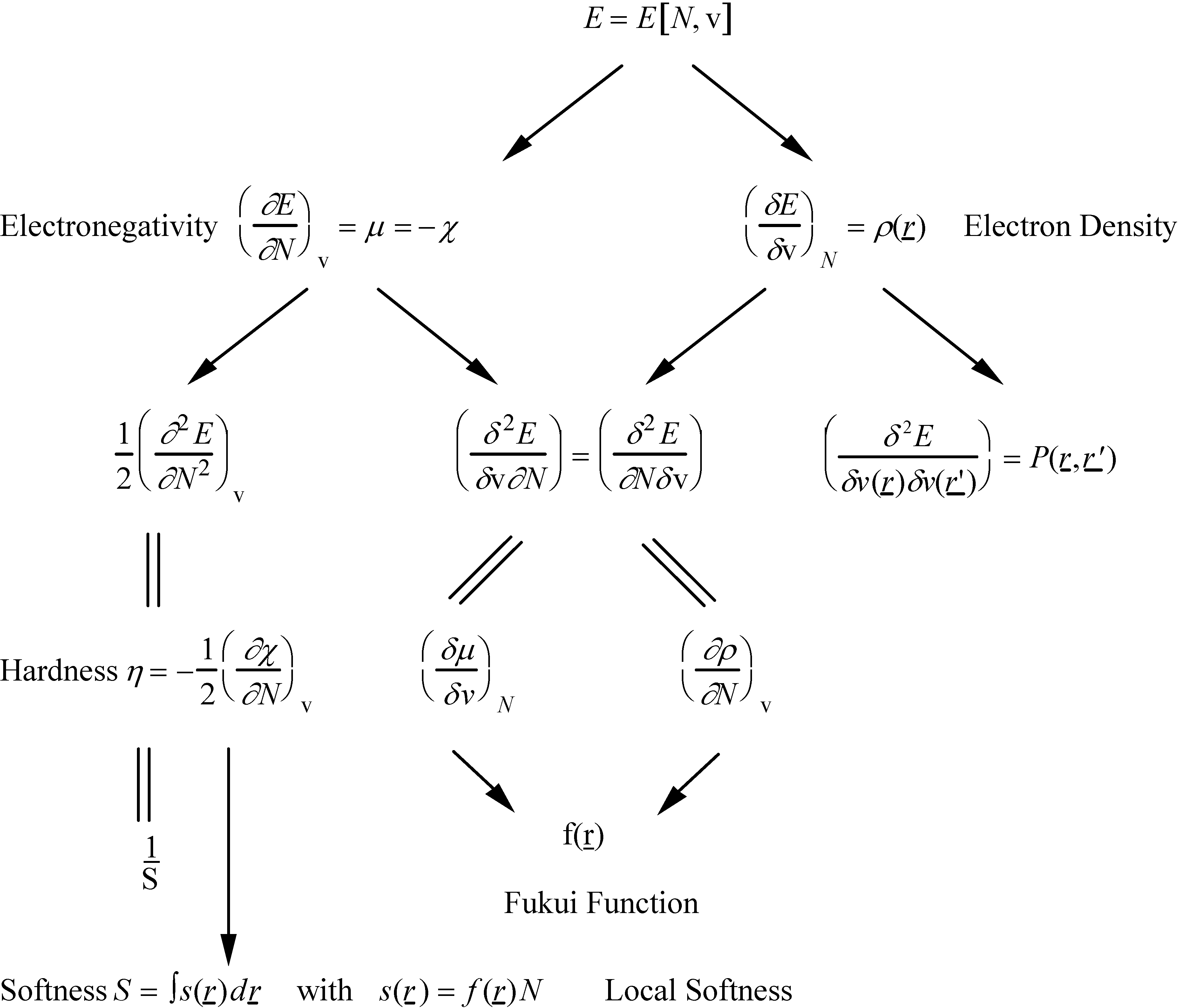
- the chemical hardness , an identification proposed by Parr and Pearson [35] for the second derivative which respect to N, , and representing the resistance of a system to changes in its number of electrons. The chemical softness S is naturally defined as the inverse ofThe analogue of equation (9) turned out to be
- -
- -
- a local version of S, s(r), obtained by multiplying S and f(r), the latter function distributing the local softness over various domains in space [38]
- -
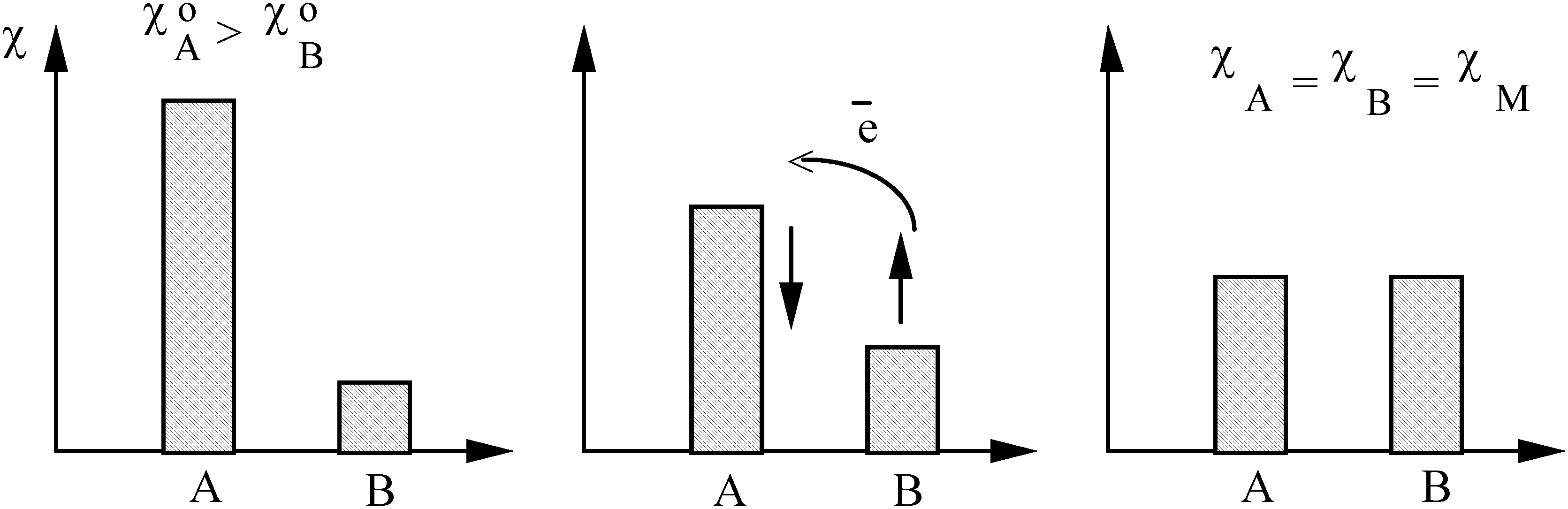
- Sanderson's electronegativity equalization principle [41,42] stating that upon molecule formation, atoms (or more general arbitrary portions of space of the reactants) with initially different electronegat combine in such a way that their "atoms-in-molecule" electronegativities are equal. The corresponding value is termed the molecular electronegativity .

- -
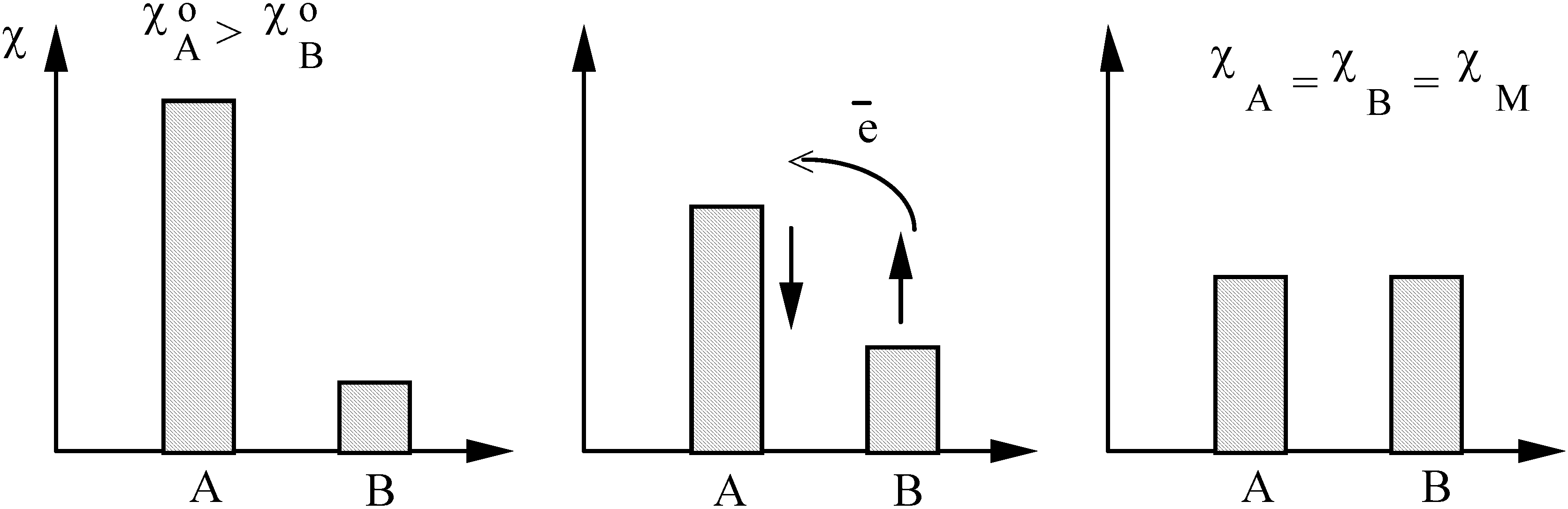
2. The structure -property (reactivity)- electron density triangle: some examples
2.1. Introduction
2.2. Organic Chemistry
2.2.1. Group properties and their use in acidity and basicity studies
- -
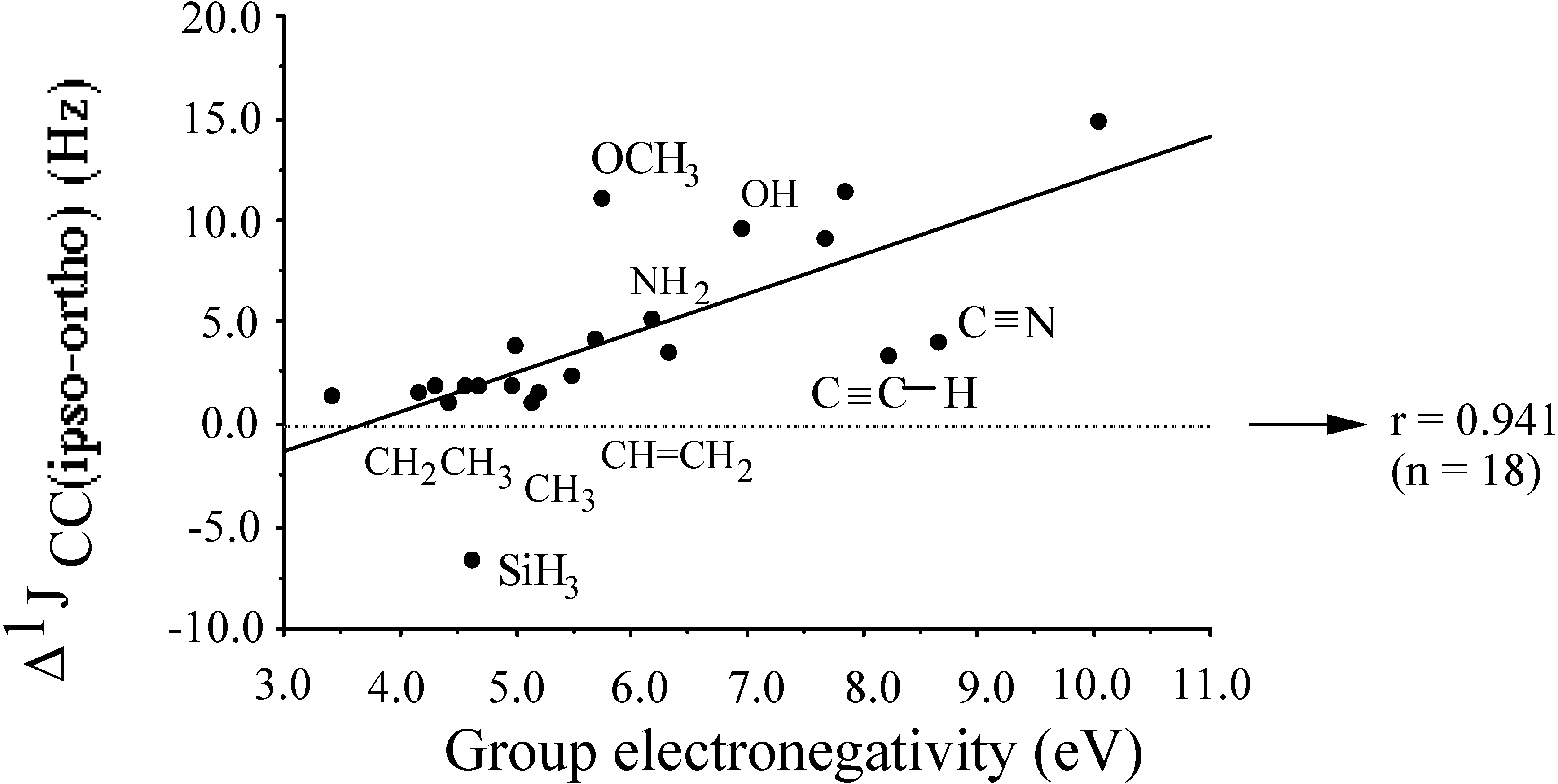
- the central atom effectχCH3 < χNH < χOH cf. χC < χN < χOindicating that upon saturation of two different atoms with hydrogens the electronegativity of the resulting groups parallels that of the naked atoms.
- -
- the second row effectχCH3 > χSiH3χNH2 > χPH2χOH > χSHshowing increasing electronegativity of a group the higher the central atom is positioned in a given column of the periodic table.
- -
- the hybridisation effectχCH2CH3 < χCH=CH2 < χCH+CHχCH2NH2 < χCH=NH2 < χC+Nindicating increasing electronegativity upon increasing s-character of the central atom of the group.
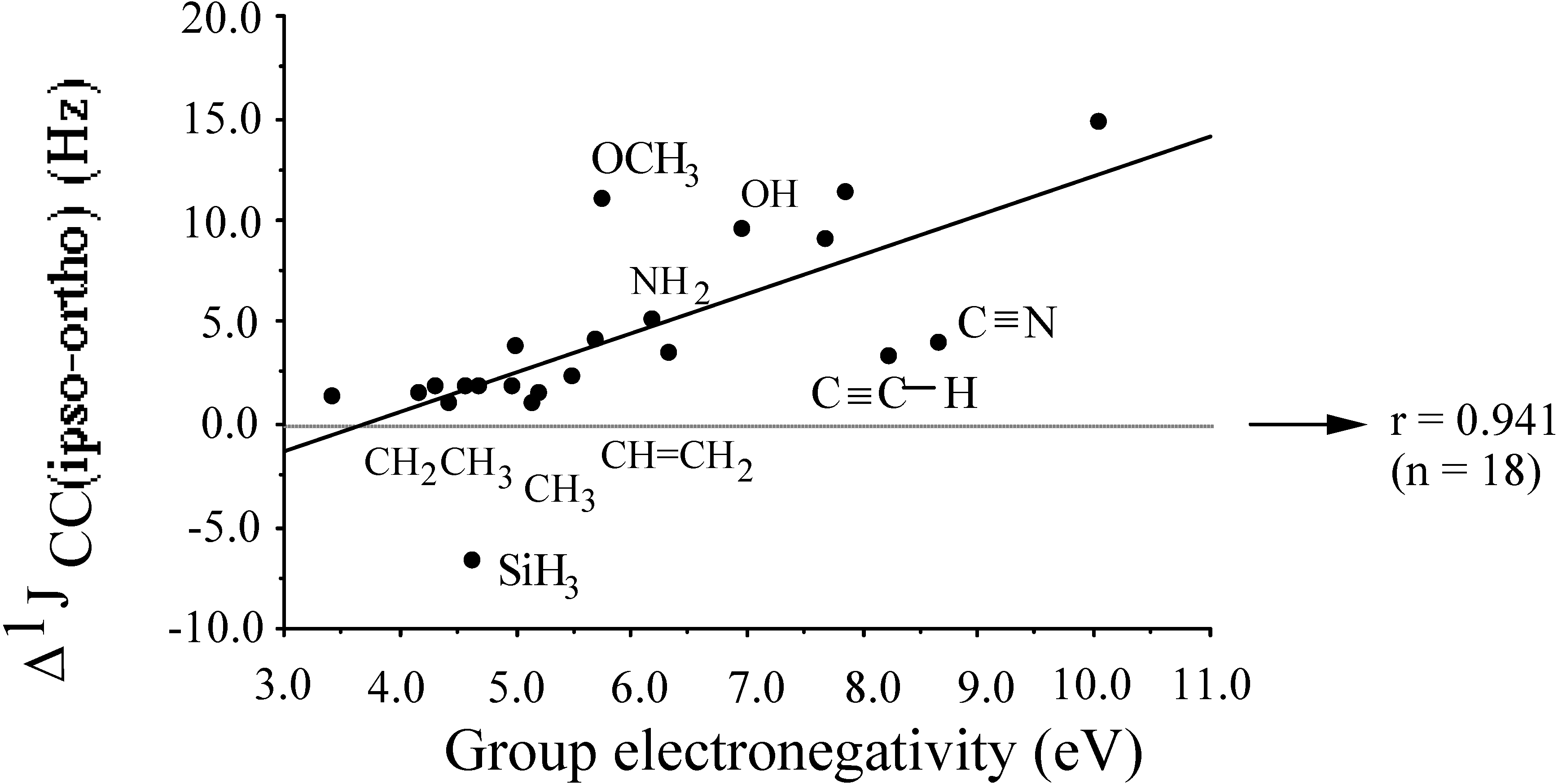
- -
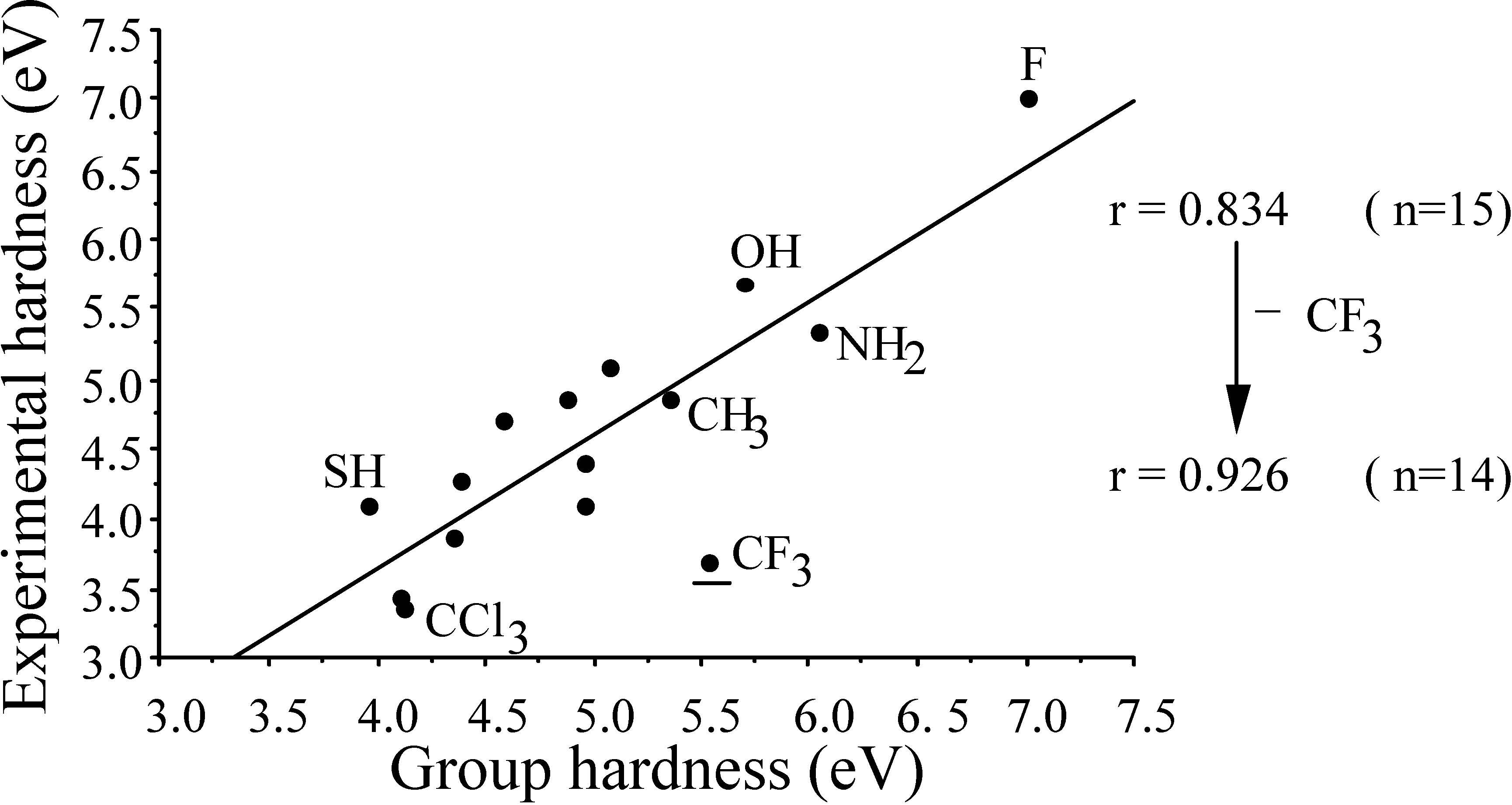
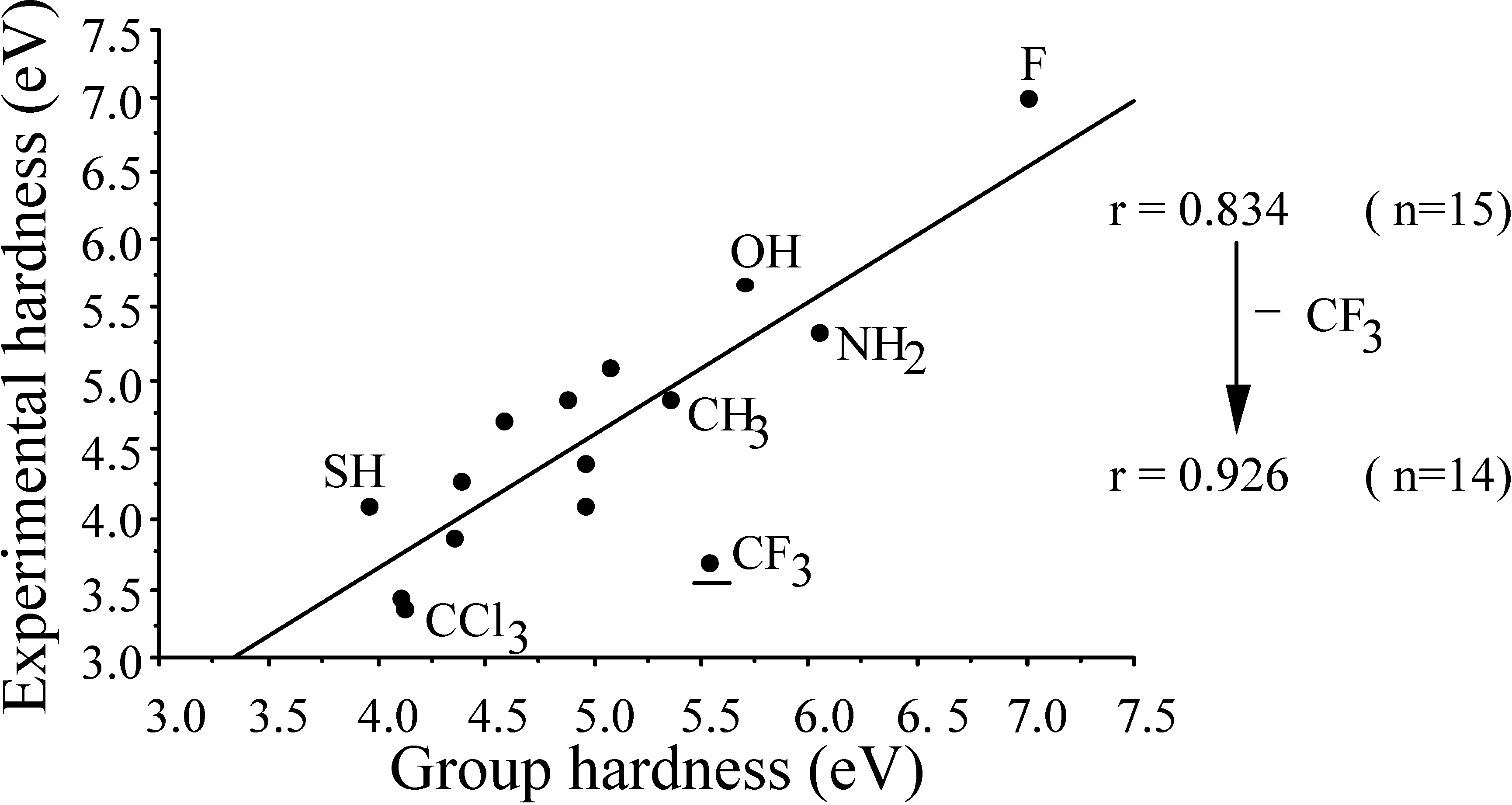
- the central atom effectχCH3 < χNH2 > ηOH < ηF cf. ηC < ηN > ηO < ηF
- -
- the second row effect (extremely important in forthcoming discussions but here already illustrating the opposite behaviour of χ and S)ηOH > ηSHηCCl3 < ηCH3 < ηCF3ηCH3 > ηSiH3
- -
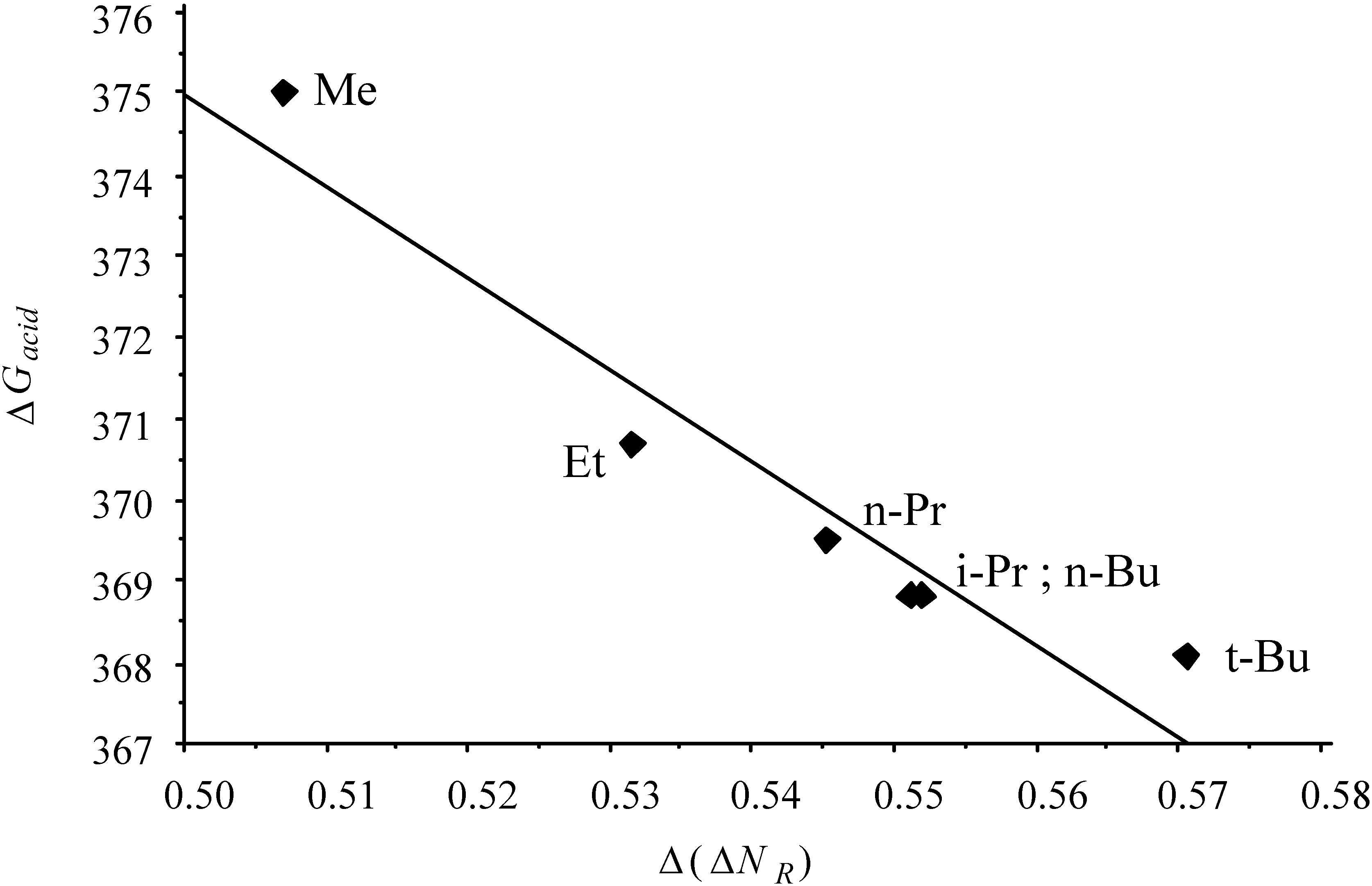
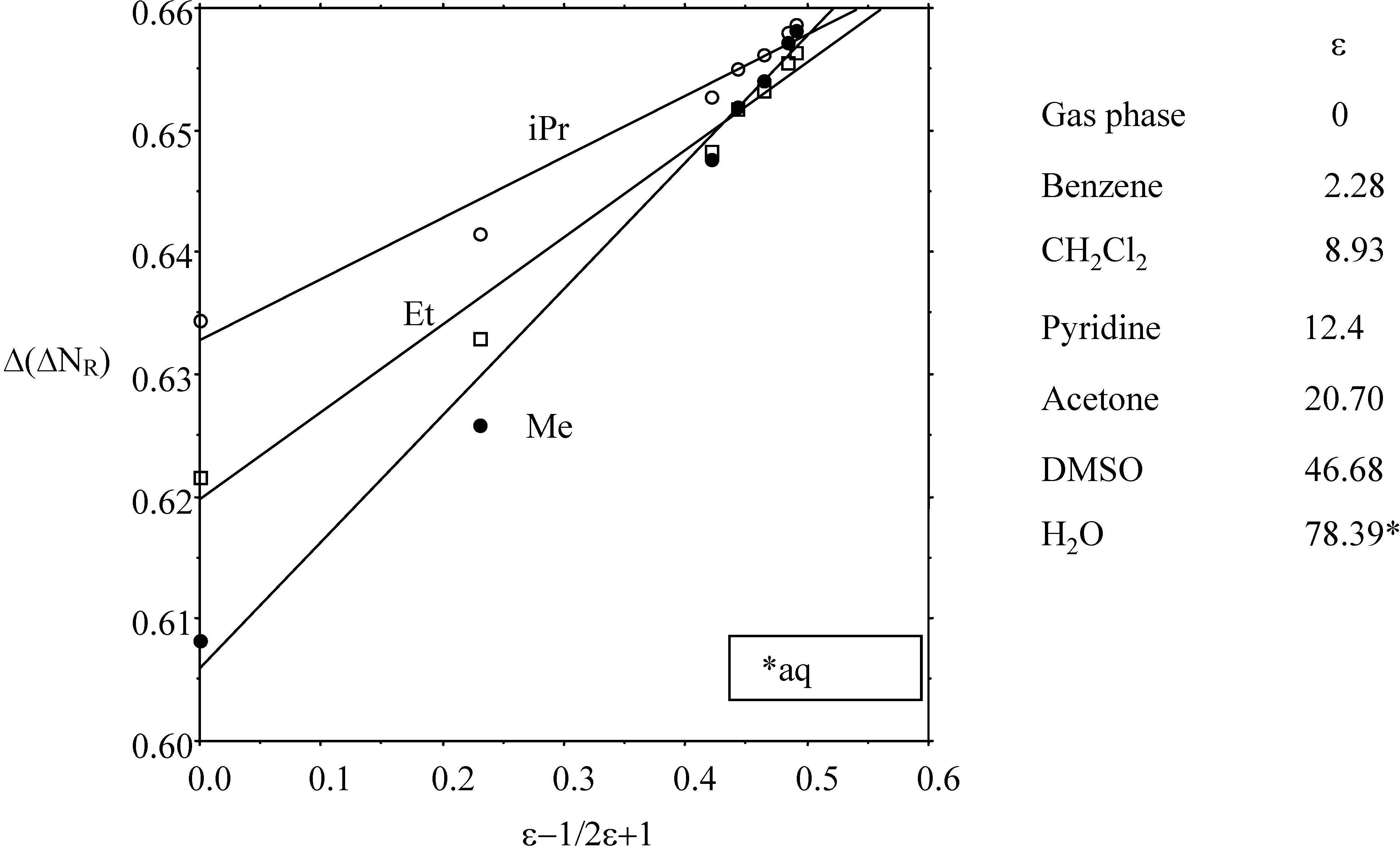
- the "volume" effect in alkyl groups, showing decreasing hardness (increasing softness) upon increasing chain length or branching. CISD η values illustrating this trend are6.60 (H) ; 5.34 (Me) ; 4.96 (Et) ; 4.70 (n-Pr) ; 4.62 (i-Pr)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| χ | η | |
| Me | 4.35 | 5.98 |
| Et | 3.76 | 5.45 |
| n-Pr | 3.68 | 5.17 |
| i-Pr | 3.44 | 5.04 |
| n-Bu | 3.57 | 5.05 |
| t-Bu | 3.30 | 4.19 |
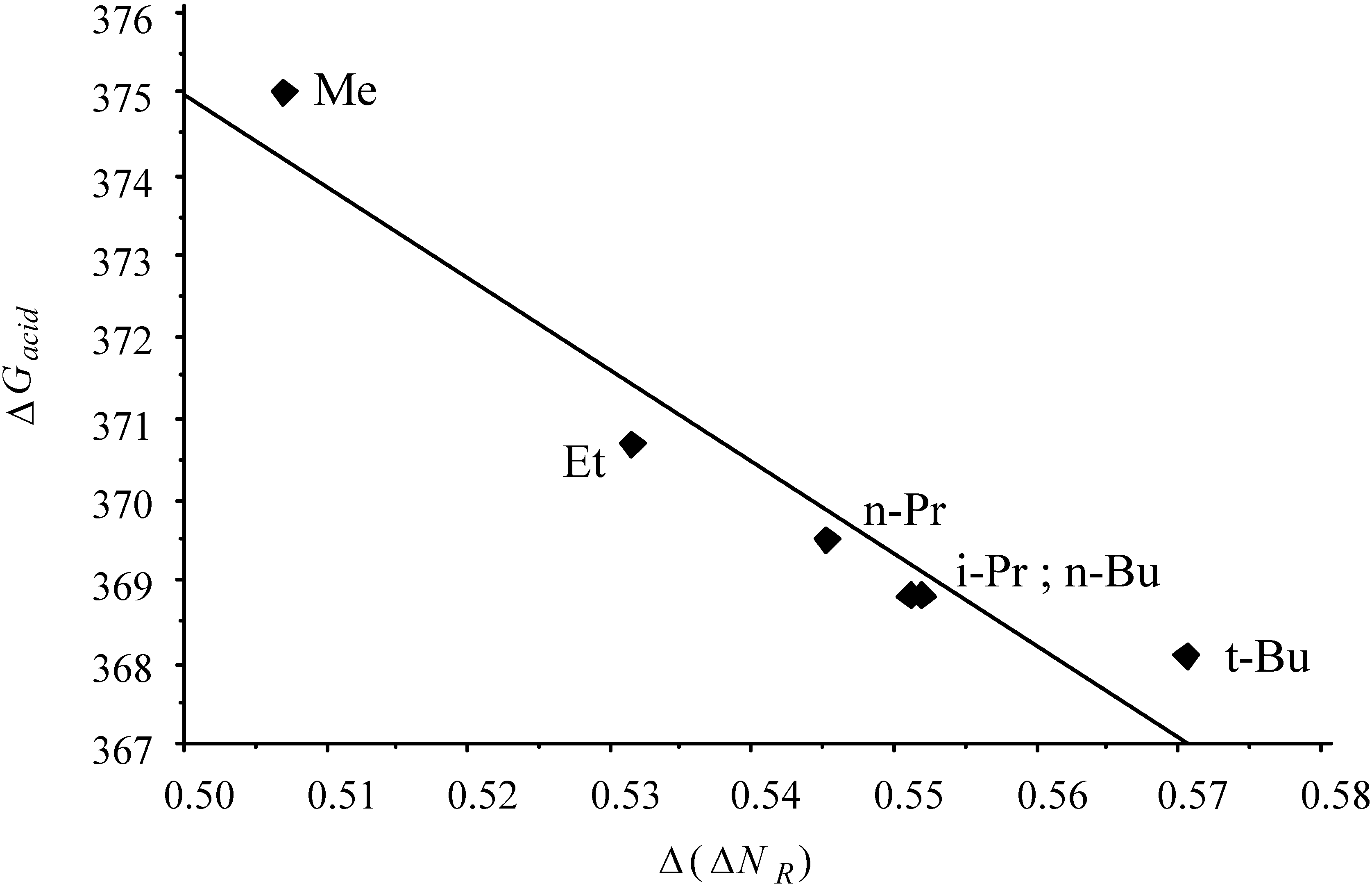
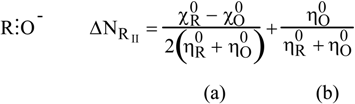
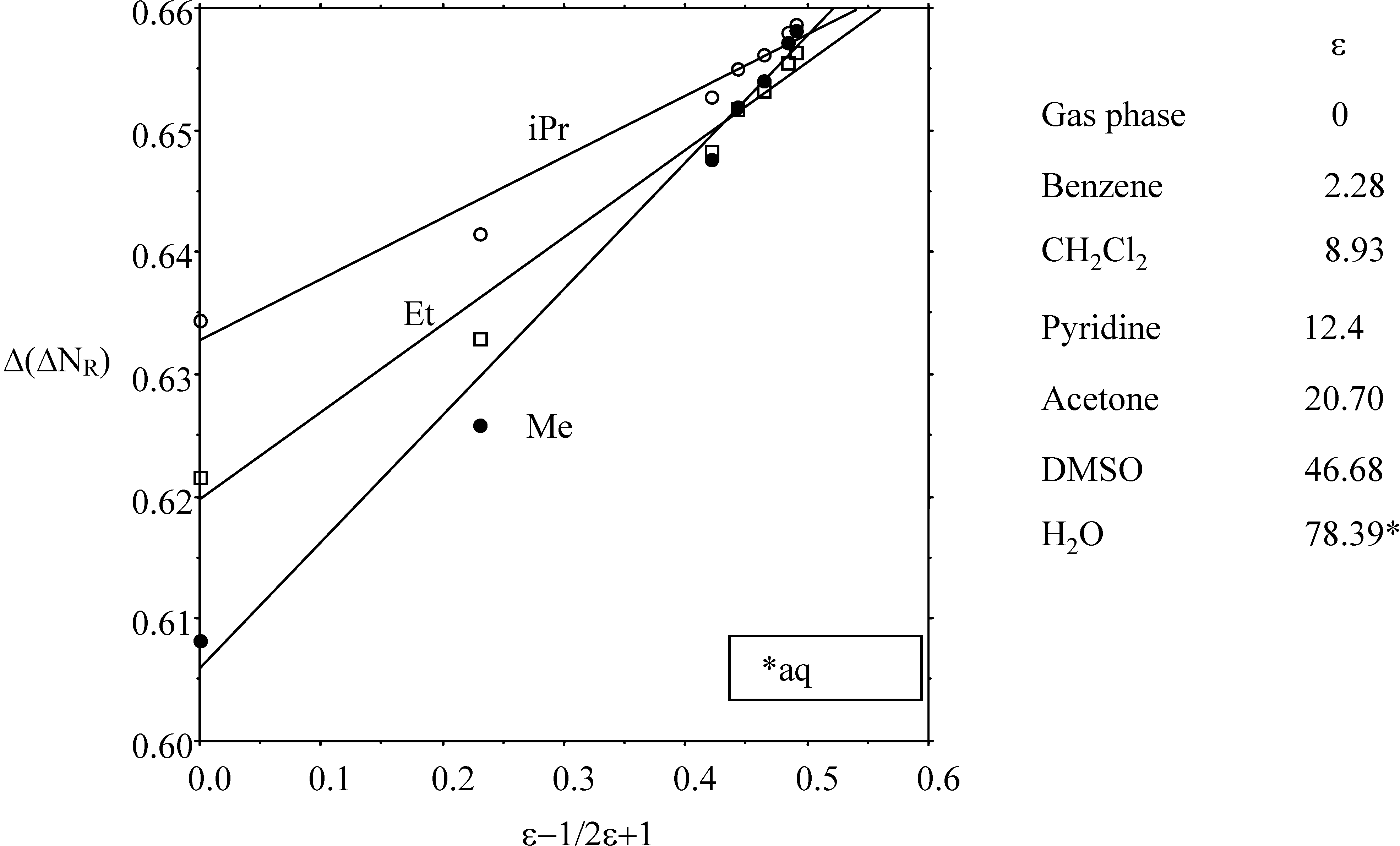
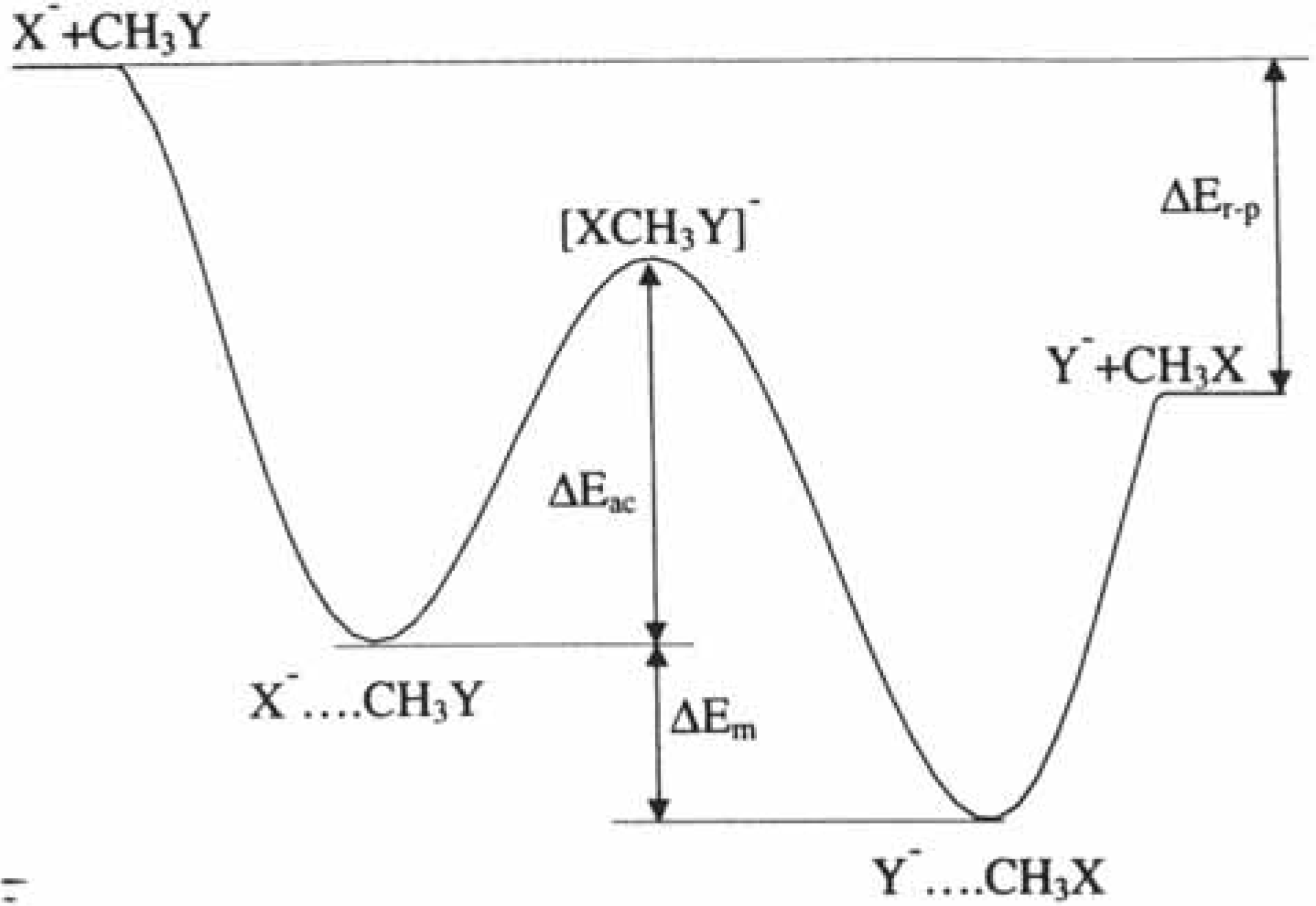
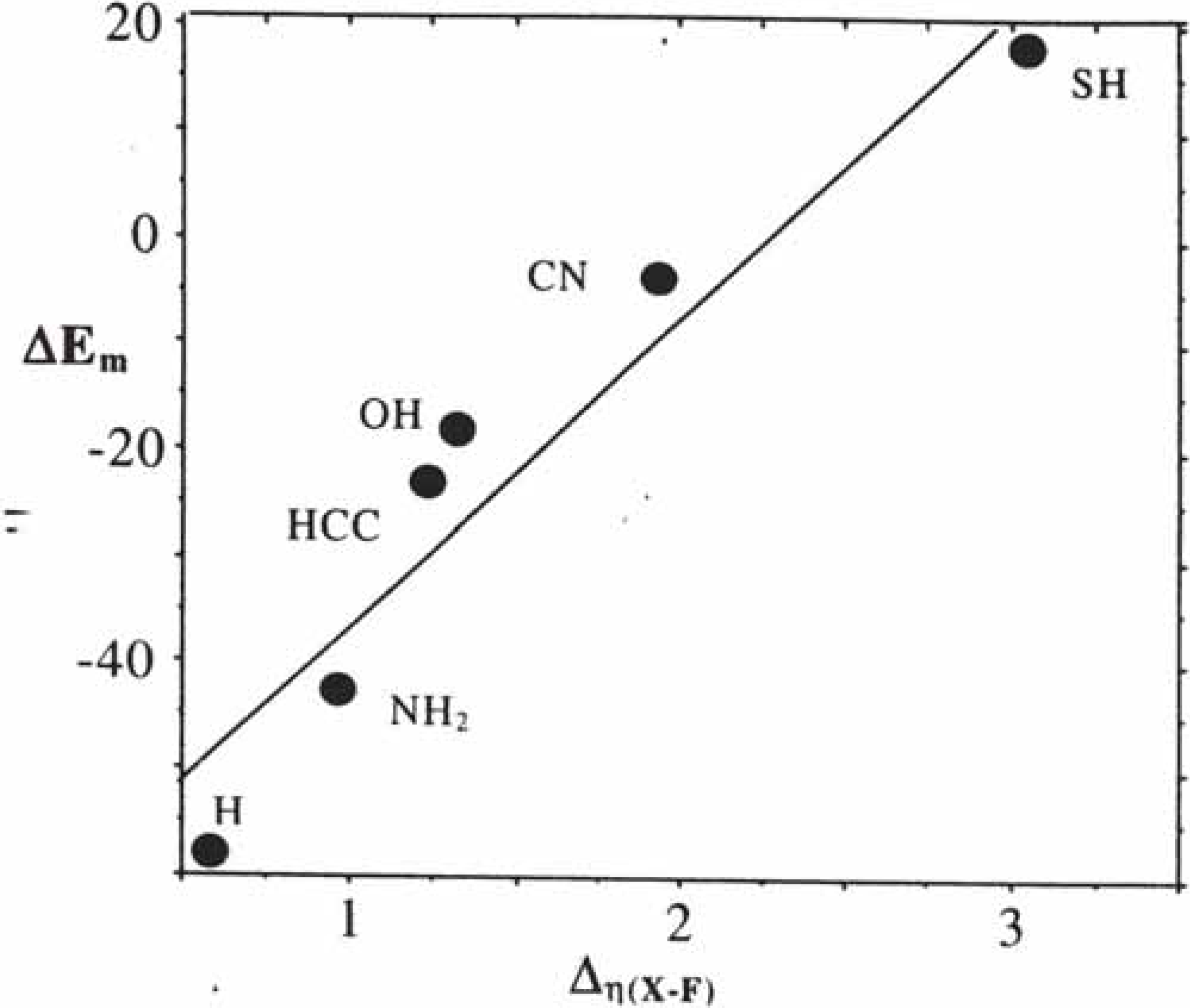
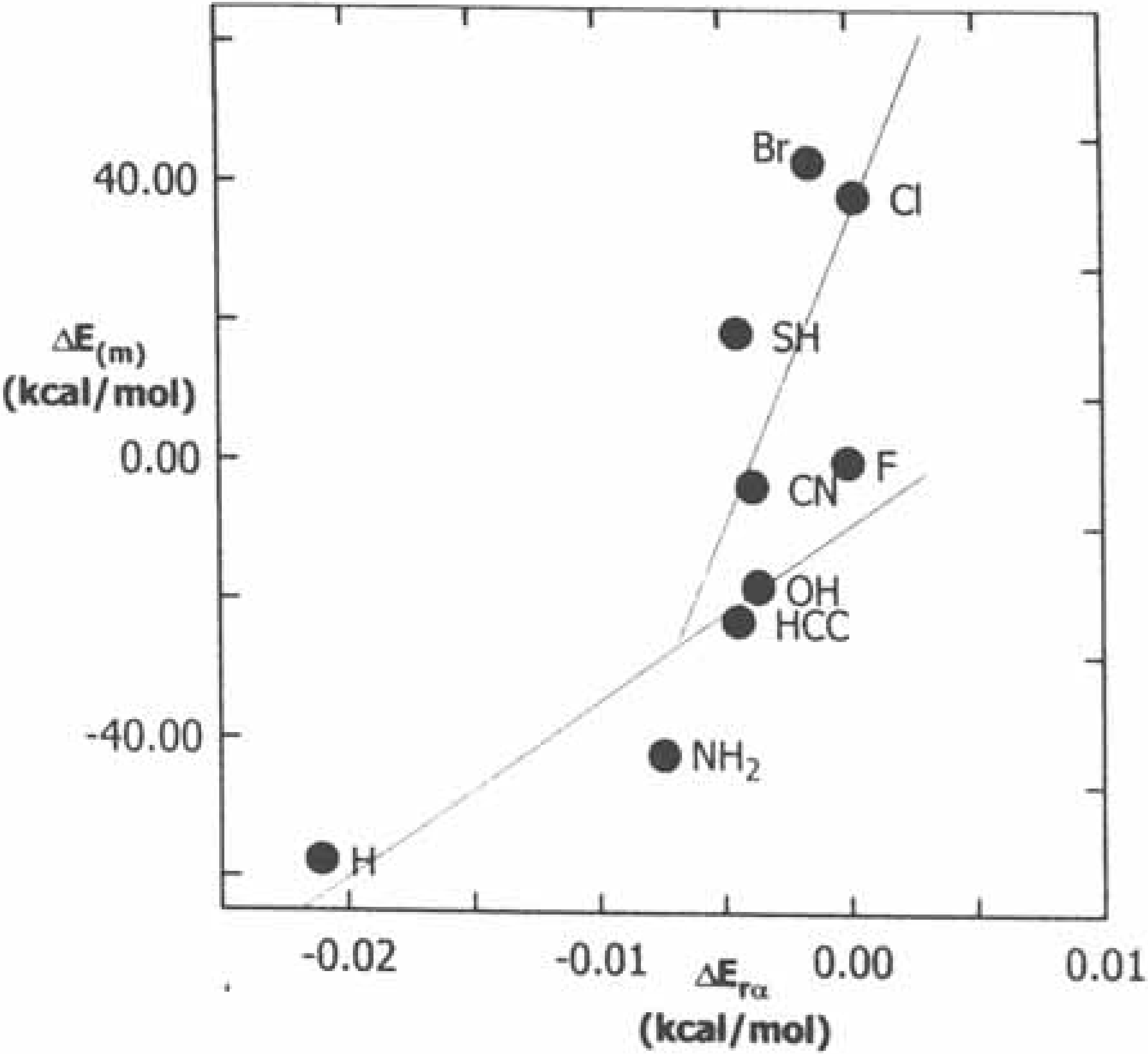
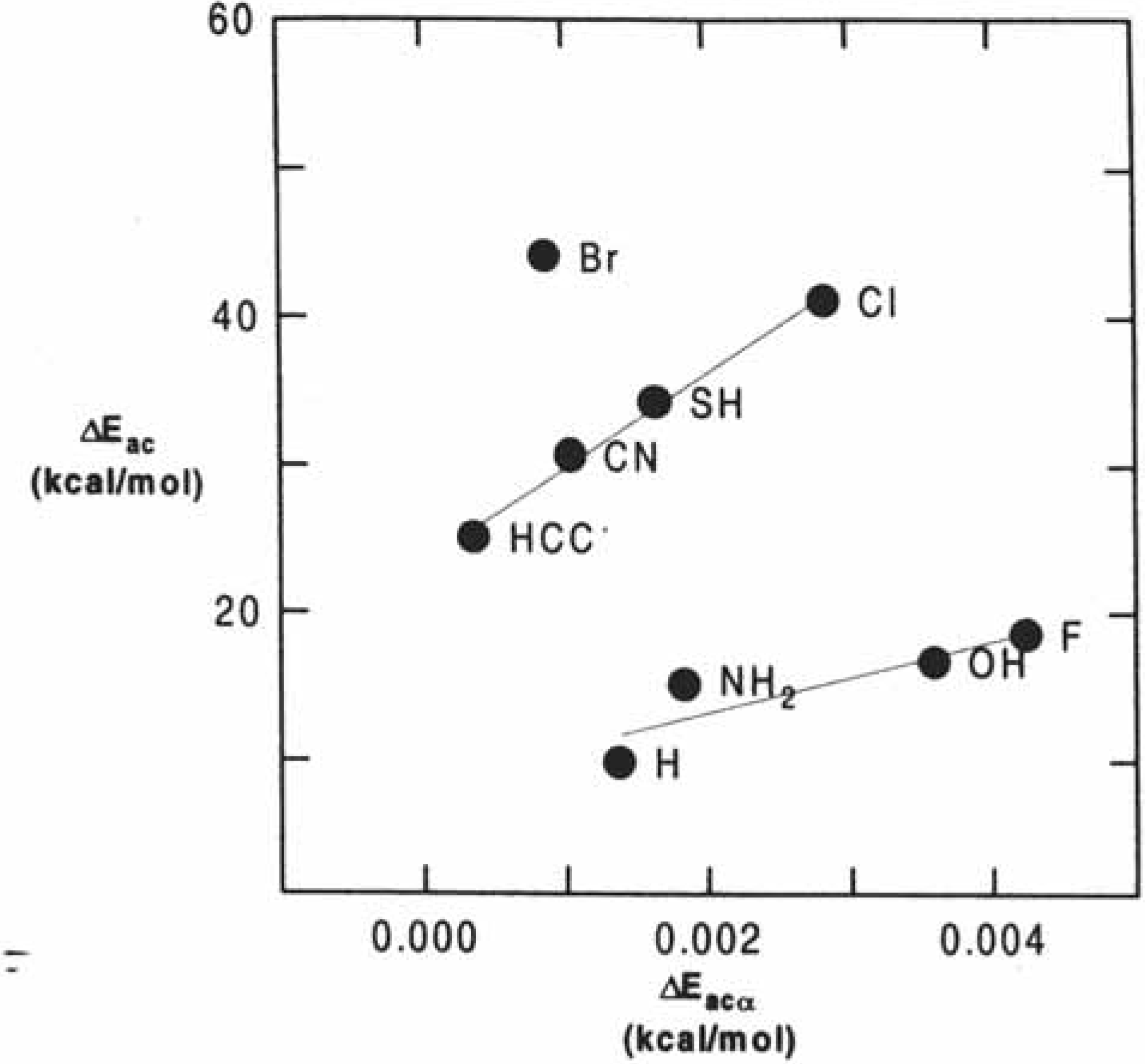
2.2.2. Kinetics of SN2 reactions
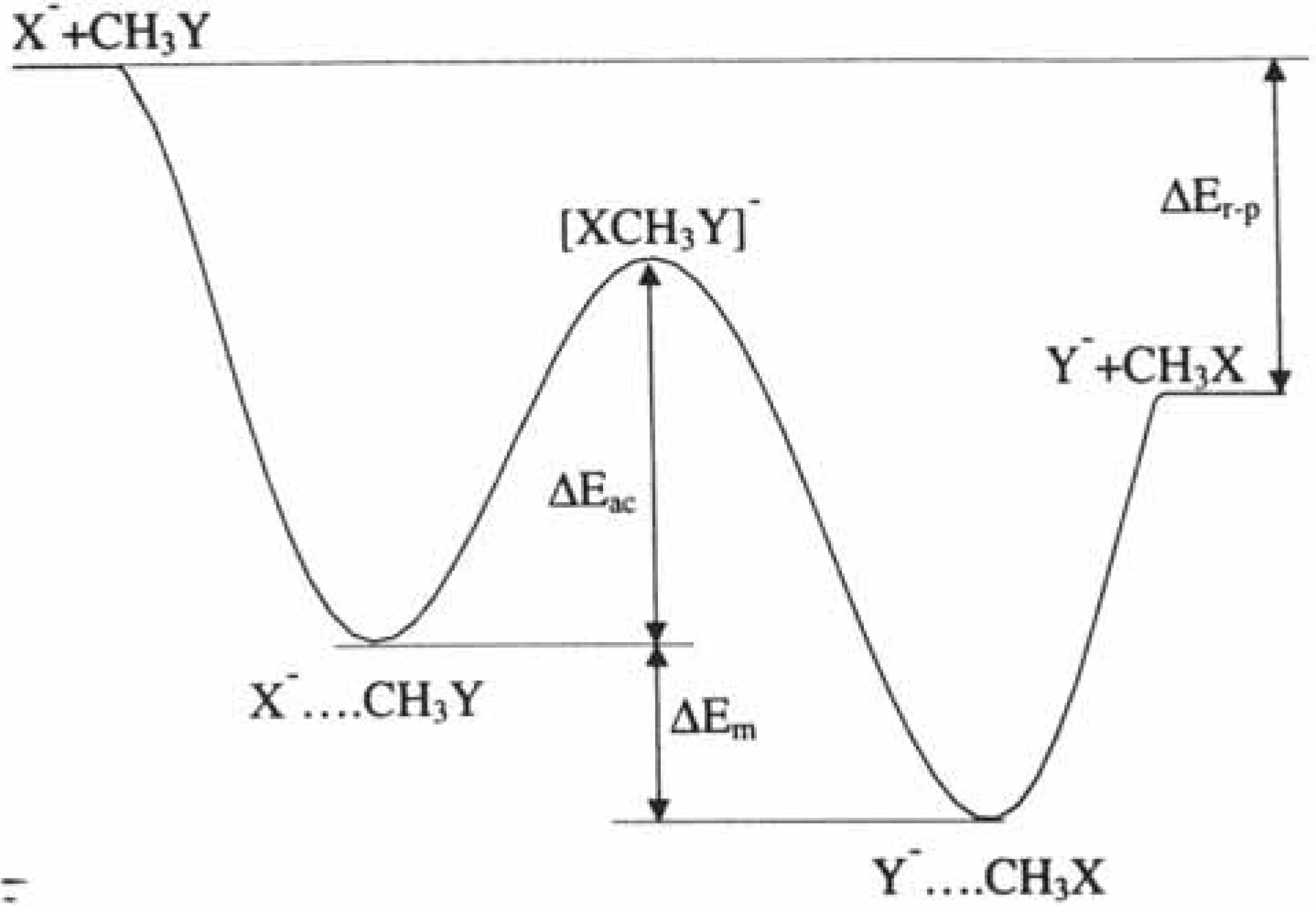
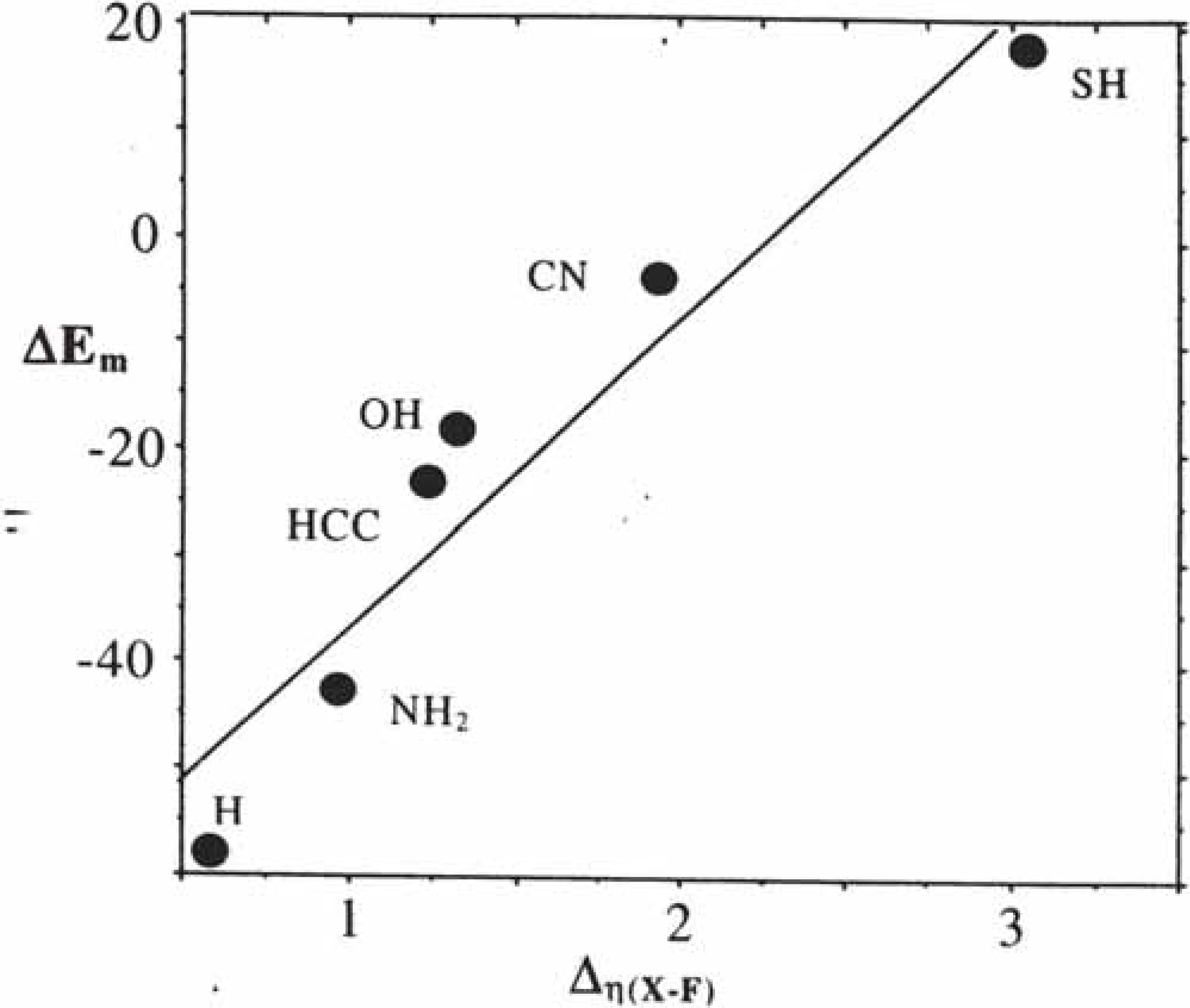


2.2.3. Regioselectivity in organic reactions: the HSAB principle

2.2.4. Similarity in Reactivity
| - CO-NH - | ||
| - CH=CH - (trans) | 0.582 | 0.576 |
| - CH=CH - (cis) | 0.506 | 0.501 |
| - CF=CH - (Z) | 0.418 | 0.635 |
| - CH2-CH2 - | 0.466 | 0.495 |
| - CH2-S - | 0.034 | 0.337 |
| - CO-CH2 - | 0.531 | 0.108 |
| - CH2-NH - | 0.412 | 0.559 |
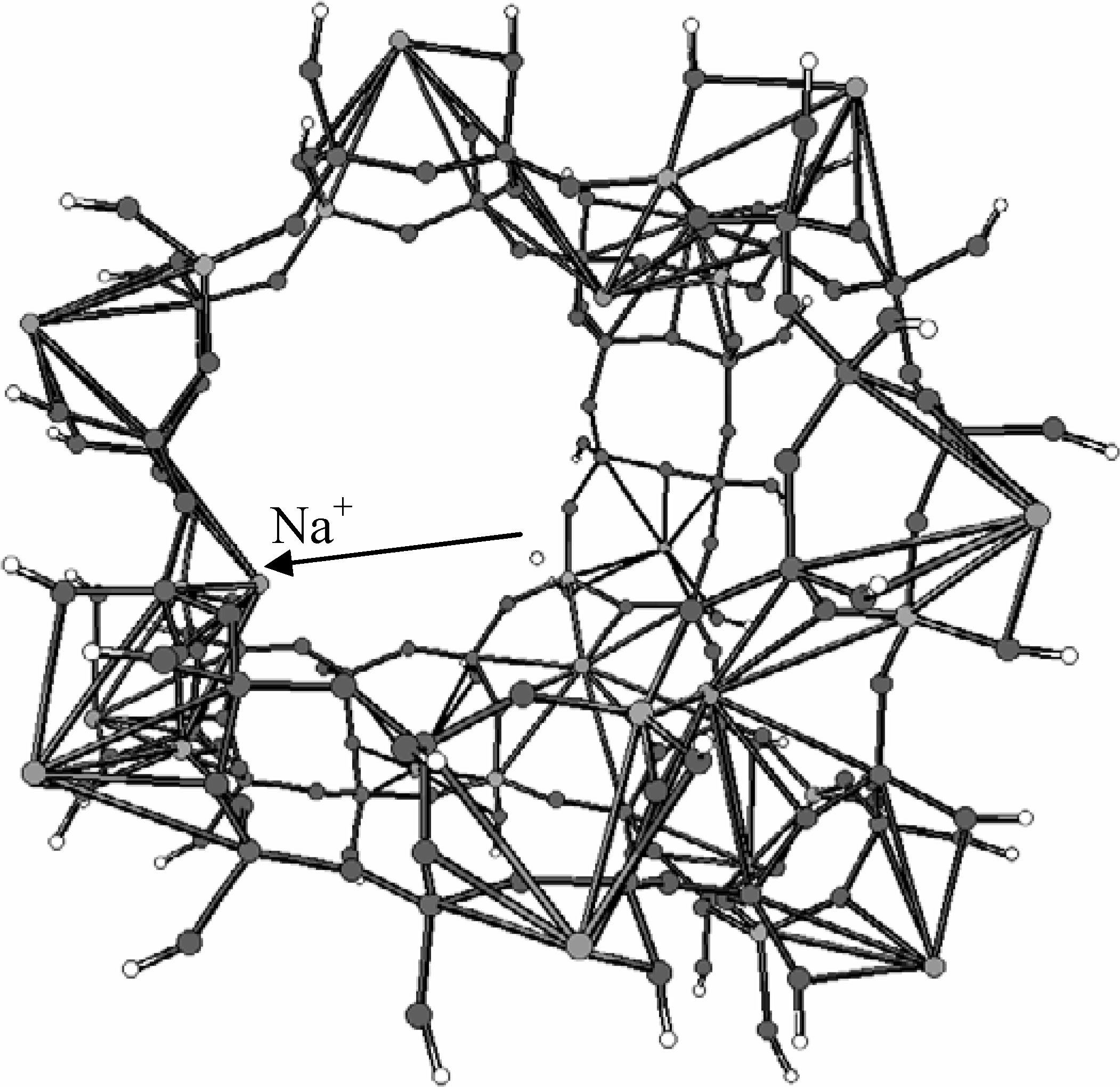
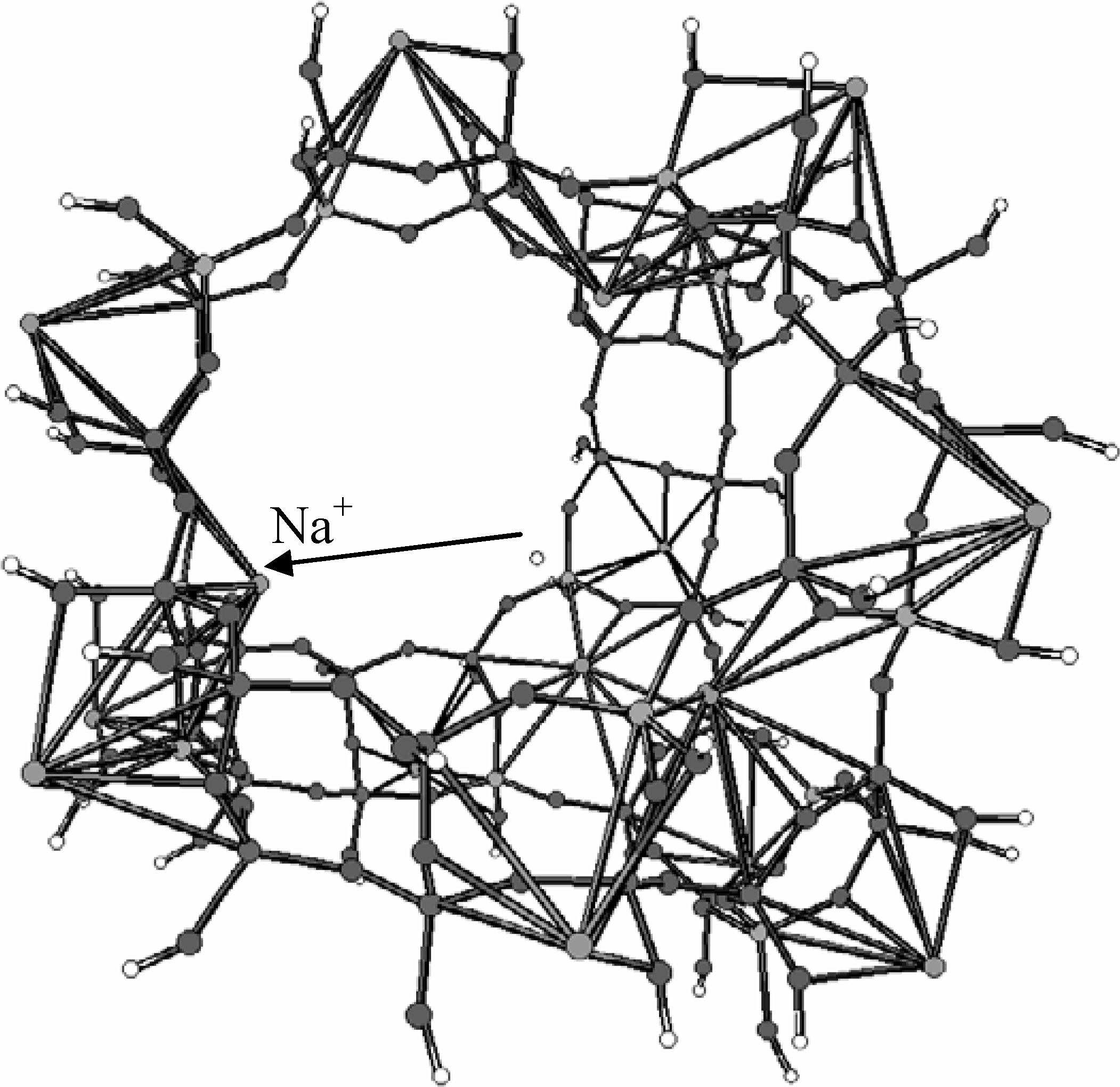
2.3. Inorganic Chemistry: Zeolites
| N2 | O2 | CO | CO2 | C2H2 | |
| K | 3.320 | 1.830 | 9.951 | 63.33 | 196.8 |
| Kexp | 31.4 | 15.4 | 85 | ||
| ΔH° | -12.9 | -7.9 | -18 | -27 | -29 |
| ΔHexp | -14 | -9.4 | -20 | -37 |
| N2/O2 | CO/N2 | CO2/CO | |
| α | 1.81 | 2.99 | 6.36 |
| αexp | 2.04 | 2.70 |
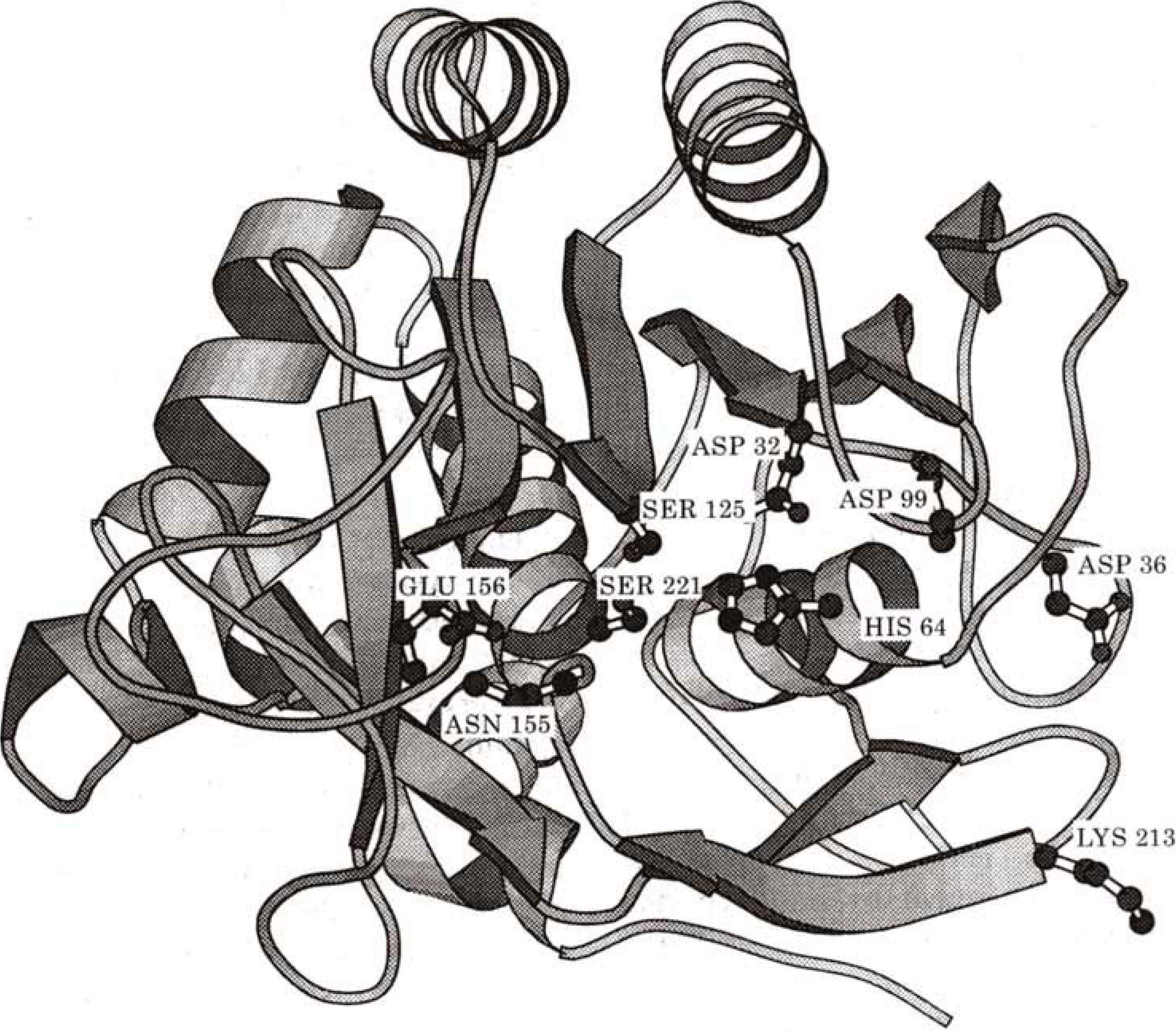
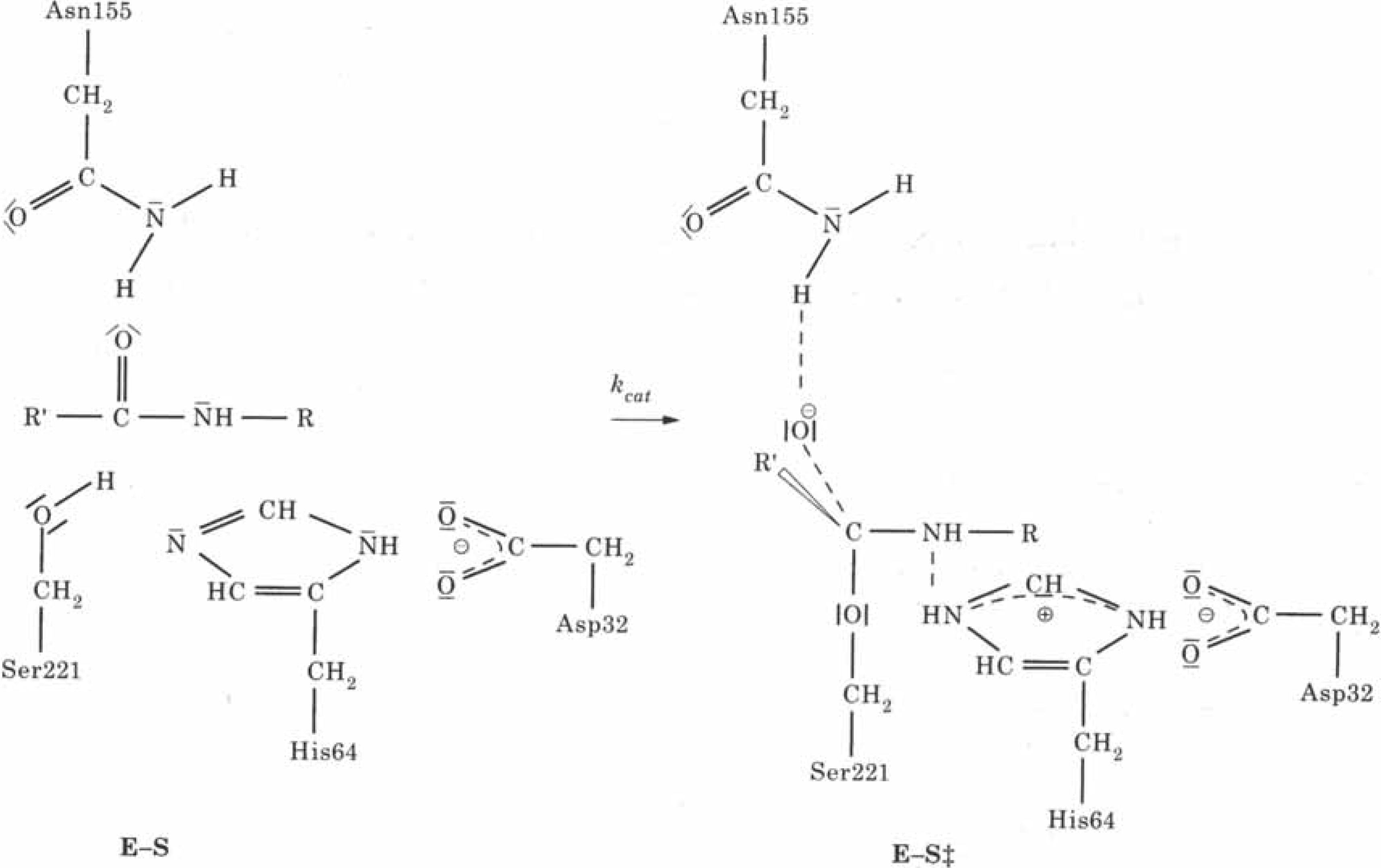
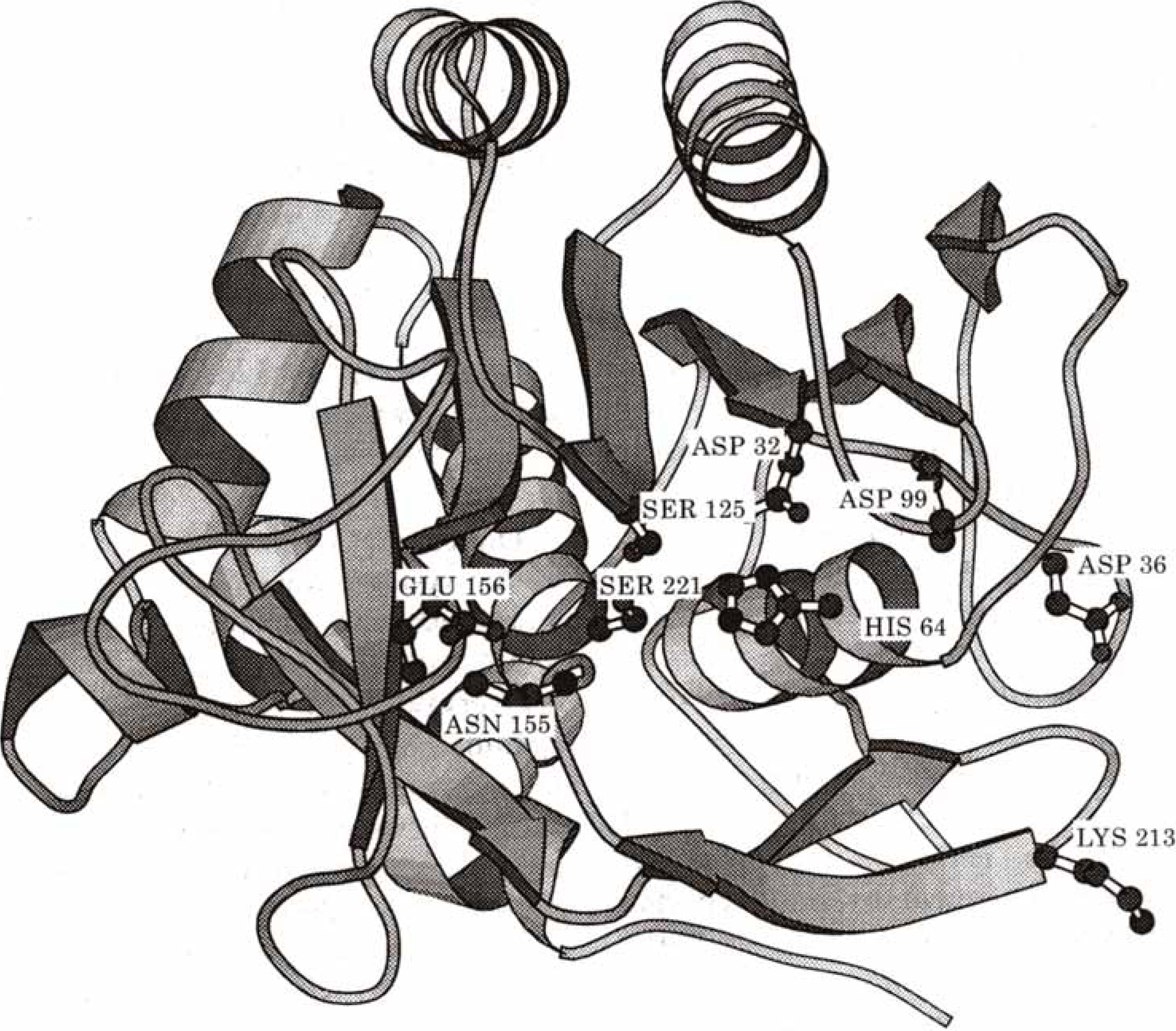
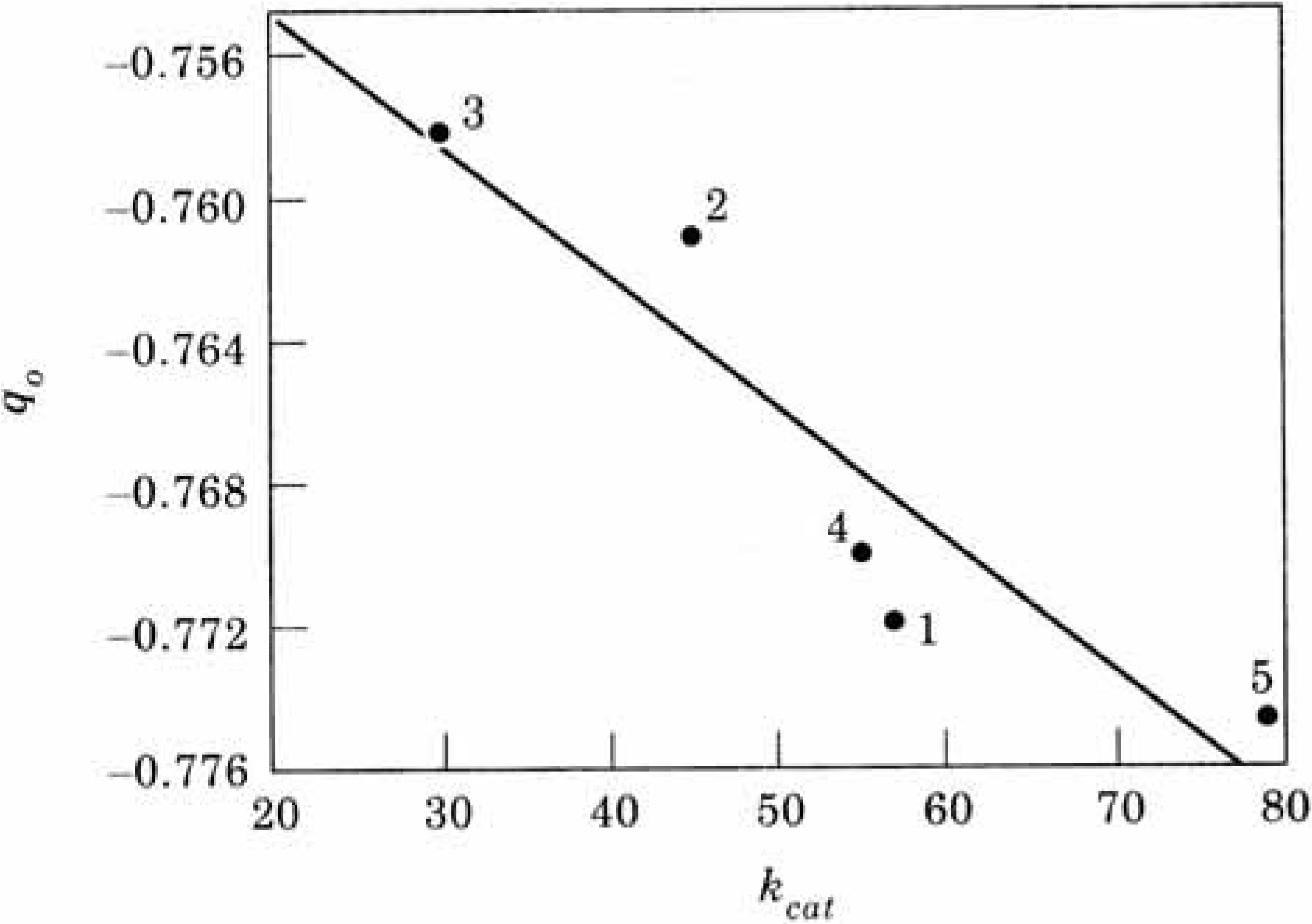
2.4. Biochemistry : Influence of point mutations on the catalytic activity of subtilisin
| qO | V(R)Min | |
| Wild Type | -0.7719 | -121.72 |
| Asp32 Ala | -0.7440 | -75.64 |
| His64 Ala | -0.7665 | -113.13 |
| Asp32 Ala : His64 Ala | -0.7487 | -67.92 |
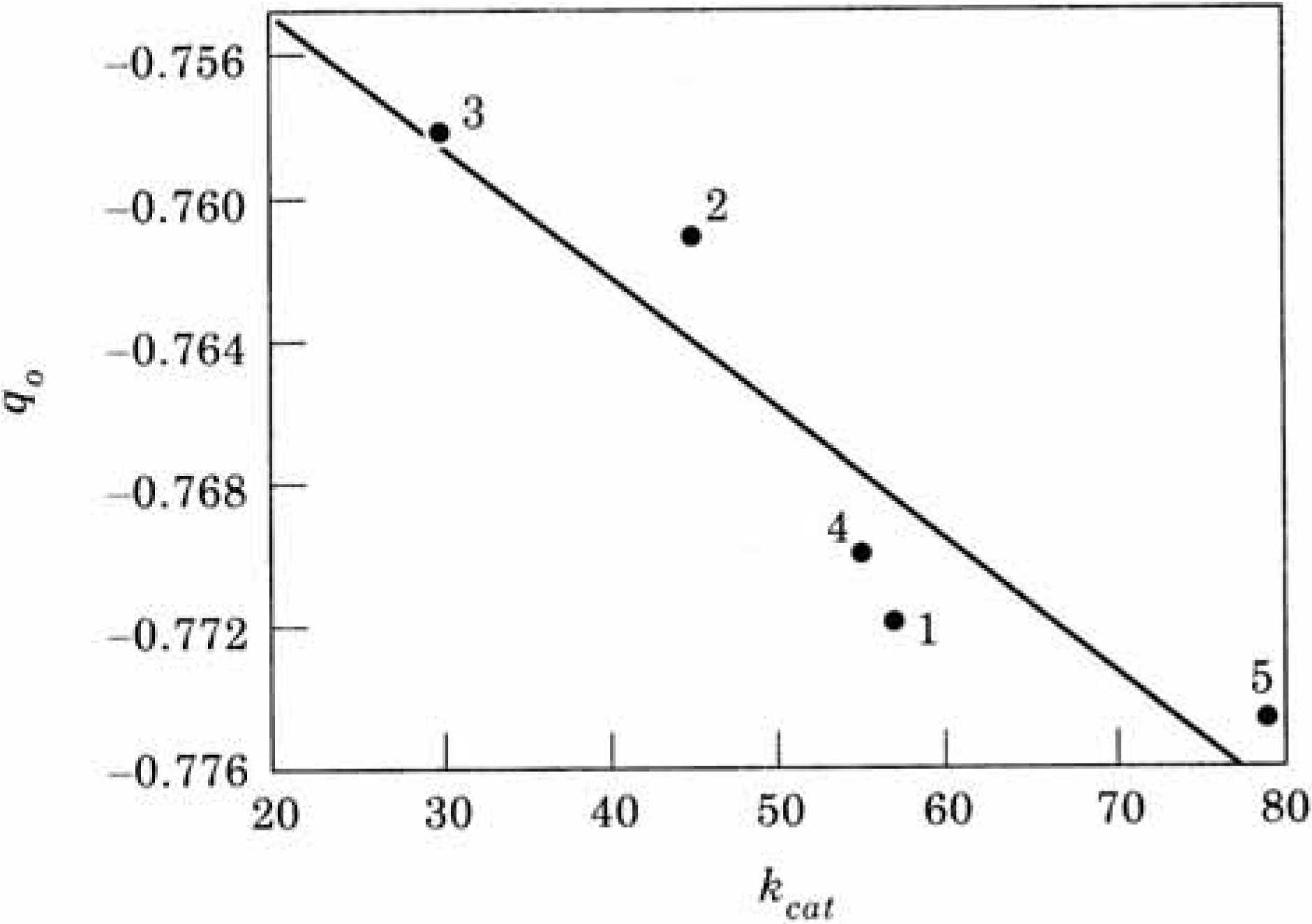
| kcat | qO | V(R)min | |
| 1. Wild Type | 57 | -0.7719 | -121.72 |
| 2. Asp99 Ser | 45 | -0.7610 | -121.33 |
| 3. Asp99 Lys | 30 | -0.7581 | -121.30 |
| 4. Glu156 Ser | 55 | -0.7700 | -122.96 |
| 5. Glu156 Lys | 79 | -0.7748 | -124.57 |
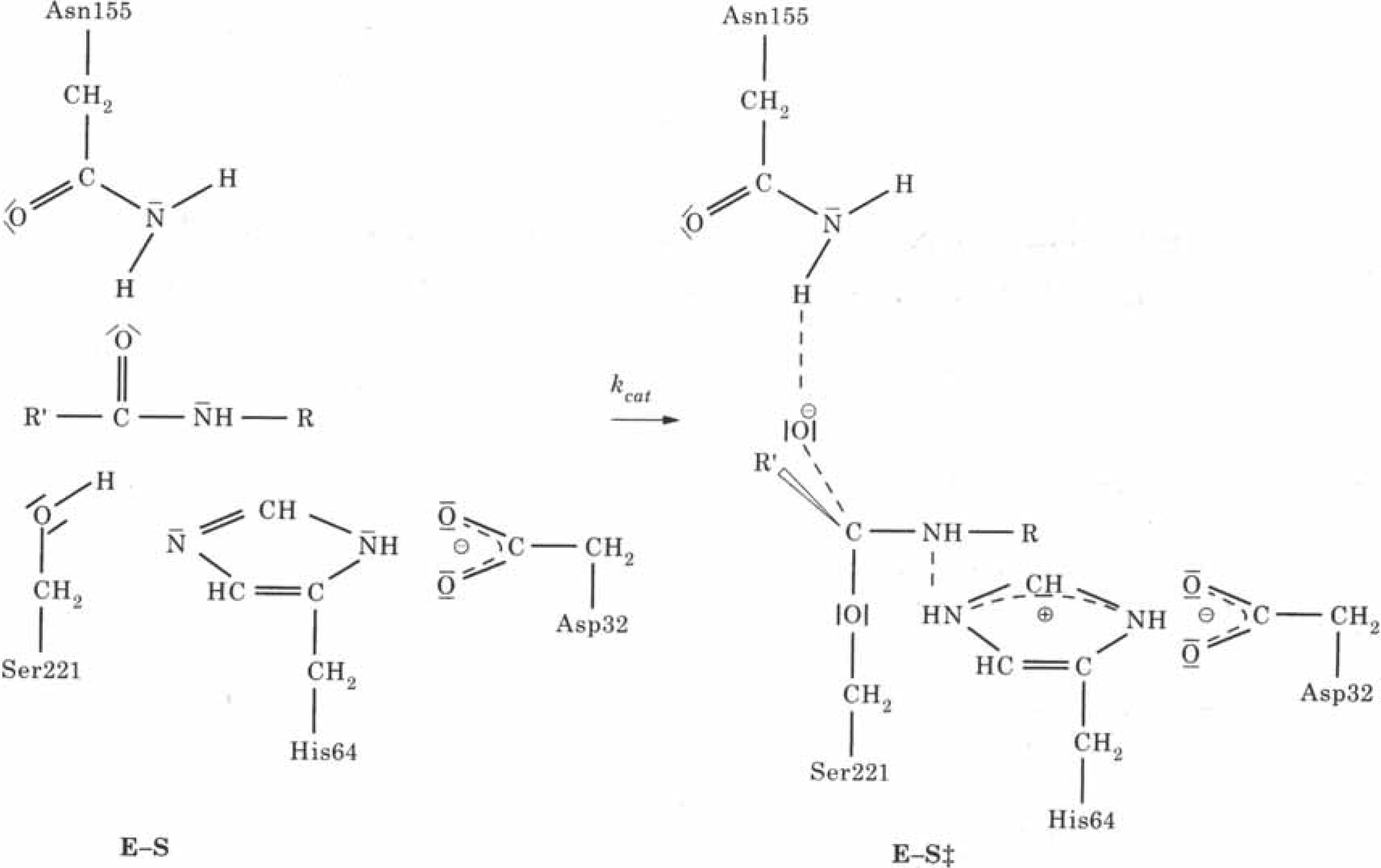
3. Conclusions
4. Acknowledgements
5. References
- Dirac, P.A.M. Proc. Roy.Soc.(London) 1929, 123, 714. [CrossRef]
- Heitler, W.; London, F. Z. Phys. 1927, 44, 455. [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, Third Edition ed; Cornell University Press: Ithaca, 1960. [Google Scholar]
- For a classic and authoritative account of Hückel's approach and its applications see A Streitwieser, A. Molecular Orbital Theory for Organic Chemistry; John Wiley, 1961. [Google Scholar]
- Mc Weeny, R. Coulson's Valence, Third Edition ed; Oxford, 1979. [Google Scholar]
- Hund, F. Z. Phys. 1928, 51, 759.Mulliken, R. S. Phys Rev. 1928, 32, 186.
- Roothaan, C. C. J. Rev. Mod. Phys. 1951, 23, 69. [CrossRef]
- For an overview see (a)Hartree, D.R. The Calculation of Atomic Structures; John Wiley: New York, 1957. [Google Scholar](b)Hehre, W. J.; Radom, L.; Schleyer, P. v. R; Pople, J. A. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986. [Google Scholar]
- MØller, C.; Plesset, M.S. Phys. Rev. 1934, 46, 618. [CrossRef]
- Shavitt, I. The Method of Configuration Interaction in Modern Theoretical Chemistry; Vol.3, Methods of Electronic Structure Theory; Schaefer, H.F., III, Ed.; Plenum Press: New York, 1977; p. 189. [Google Scholar]
- Bartlett, R.J. J. Phys. Chem. 1989, 93, 1697. [CrossRef]
- Pople, J.A.; et al. GAUSSIAN 98; 1998; and previous releases (GAUSSIAN 94, GAUSSIAN 92, ..., GAUSSIAN 70); Gaussian Inc.: Pittsburgh A. [Google Scholar]
- The Encyclopedia of Computational Chemistry; Schleyer, P. v. Rague; Allinger, N. L.; Clark, T.; Gasteiger, J.; Kollman, P. A.; Schaefer, H. F.; Schreiner, P. R. (Eds.) John Wiley & Sons Ltd.: Chichester, 1998.
- Pople, J.A. J. Chem. Phys. 1966, 43, S229.
- Hohenberg, P.; Kohn, W. Phys. Rev. B 1964, 136, 864.
- Mc Weeny, R.; Sutcliffe, B.T. Methods of Molecular Quantum Mechanics; Academic Press: London, 1969; Chapter 4. [Google Scholar]
- Kohn, W.; Sham, L. Phys. Rev. A 1965, 140, 1133.
- Parr, R.G.; Yang, W. Ann. Rev. Phys. Chem. 1995, 46, 701.
- Becke, A.D. J. Chem. Phys 1993, 98, 5648.Lee, C.; Yang, W.; Parr, R.G. Phys. Rev. B 1998, 37, 785.
- Geerlings, P.; De Proft, F.; Langenaeker, W. Adv. Quantum Chem. 1999, 33, 303.
- Kohn, W.; Becke, A.; Parr, R.G. J. Phys. Chem. 1996, 100, 12974. [CrossRef]
- Koch, W.; Holthausen, M.C. A Chemistry's Guide to Density Functional Theory; Wiley-VCH: Weinheim, 2000. [Google Scholar]
- Parr, R.G. Density Functional Methods in Physics; Dreizler, R.M., da Providencia, J., Eds.; Plenum, 1985; p. 141. [Google Scholar]
- England, W.; Salmon, L.S.; Rüdenberg, K. Fortschr. Chem. Forsch. 1971, 23, 3, and references therein.
- Foster, M.; Boys, S.F. Rev. Mod. Phys. 1960, 32, 300. [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden : a pre- and post-processing program for molecular and electronic structures. J. Comp.-Aided Mol. Design 2000, 14, 123. [Google Scholar] [CrossRef]
- Müller, N.; Pritchard, D.E. J. Chem. Phys. 1959, 31, 768.
- Figeys, H. P.; Geerlings, P.; Raeymaekers, P.; Van Lommen, G.; Defay, N. Tetrahedron 1975, 31, 1731.
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford, 1989; Chapter. [Google Scholar]
- Parr, R.G.; Donelly, R.A.; Levy, M.; Palke, W.E. J. Chem. Phys. 1978, 68, 3801. [CrossRef]
- Iczkowski, R.P.; Margrave, J.L. J. Am. Chem. Soc. 1961, 83, 3547. [CrossRef]
- Mulliken, R.S. J. Chem. Phys. 1934, 2, 782.
- For a series of papers covering various aspects of electronegativity see : Structure and Bonding; Sen, K.D.; JØrgensen, C.K. (Eds.) Springer Verlag: Berlin, 1987; Vol.66.
- Nalewajski, R.F.; Parr, R.G. J. Chem. Phys. 1982, 77, 399.
- Parr, R.G.; Pearson, R.G. J. Am. Chem. Soc. 1983, 105, 7512. [CrossRef]
- Parr, R.G.; Yang, W. J. Am. Chem. Soc. 1984, 106, 4049. [CrossRef]
- Fukui, K.; Yonezawa, T.; Shinghu, H. J. Chem. Phys. 1952, 20, 722.
- Yang, W.; Parr, R.G. Proc. Natl. Acad. Sci. 1985, 82, 6723.
- Chermette, H. J. Comp. Chem. 1999, 20, 129.
- De Proft, F.; Geerlings, P. Chem. Rev. 2001, 101, 1451.Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. submitted.
- Sanderson, R.T. Science 1955, 121, 207.
- Sanderson, R.T. Polar Covalence; Academic Press: New York, 1983. [Google Scholar]
- Pearson, R.G. J. Am. Chem. Soc. 1963, 85, 3533. [CrossRef]
- Pearson, R.G. Chemical Hardness; J. Wiley: New York, 1997. [Google Scholar]
- Chattaraj, P.K.; Lee, P.K.; Parr, R.G. J. Am. Chem. Soc. 1991, 113, 1855. [CrossRef]
- Parr, R.G.; Chattaraj, P.K. J. Am. Chem. Soc. 1991, 113, 1854. [CrossRef]
- De Proft, F.; Martin, J.M.L.; Geerlings, P. Chem. Phys. Lett. 1996, 250, 393, 1996, 256, 400.
- De Proft, F.; Geerlings, P. J. Chem. Phys. 1997, 106, 3270.
- De Oliveira, G.; Martin, J.M.L.; De Proft, F.; Geerlings, P. Phys. Rev. 1999, A60, 1034. [CrossRef]
- See for example the impressive series The Chemistry of Functional Groups; Patai, S. (Ed.) Interscience Publishers: London.
- De Proft, F.; Langenaeker, W.; Geerlings, P. J. Phys. Chem. 1993, 97, 1826. [CrossRef]
- Datta, D.; Nabakishwar, S.S. J. Phys. Chem. 1990, 94, 2184.
- Wray, V.; Ernst, L.; Luna, T.; Jakobsen, H.-J. J. Magn. Res. 1980, 40, 55.
- Pearson, R.G. J. Am. Chem. Soc. 1998, 110, 7684. [CrossRef]
- De Proft, F.; Langenaeker, W.; Geerlings, P. Tetrahedron 1995, 55, 4021. [CrossRef]
- Lias, S.G.; Bartmess, J.-E.; Liebman, J.F.; Holmes, J.L.; Levin, R.D.; Mallard, W.G. J. Phys. Chem. Ref. Data 1988, 17 (Suppl 1).
- Bartmess, J.E.; Scott, J.A.; Mc Iver Jr., R.T. J. Am. Chem. Soc. 1979, 101, 6056. [CrossRef]
- Taylor, R. Electrophilic Aromatic Substitution; J. Wiley: New York, 1990. [Google Scholar]
- Vanermen, G.; Toppet, S.; Van Beylen, M.; Geerlings, P. J. Chem. Soc., Perkin Transactions 2 1986, 699.
- Wiberg, K.; Keith, T.A.; Frisch, M.J.; Muacko, M. J. Phys. Chem. 1995, 99, 9072. [CrossRef]
- Safi, B.; Choho, K.; De Proft, F.; Geerlings, P. J. Phys. Chem. 1998, 5253, 5253. [CrossRef]
- Isaacs, N. Physical Organic Chemistry; Longman Scientific and Technical: Singapore, 1995. [Google Scholar]
- Shaik, S.S.; Schlegel, H.B.; Wolfe, S. Theoretical Aspect of Physical Organic Chemistry; John Wiley, 1992. [Google Scholar]
- Safi, B.; Choho, K.; Geerlings, P. J. Phys. Chem 2001, A105, 591. [CrossRef]
- Gazquez, J.L. J. Phys. Chem. 1997, A101, 8967. [CrossRef]
- Politzer, P. J. Chem. Phys. 1987, 86, 1072.
- Chandra, A. K.; Uchimaru, T. J. Phys. Chem. A 2001, 105, 3578. [CrossRef]
- Gazquez, J.L.; Mendez, F. J. Phys. Chem. 1994, 98, 459.Damoun, S.; Van de Woude, G.; Mendez, F.; Geerlings, P. J. Phys. Chem. 1997, A101, 886.
- Geerlings, P.; De Proft, F. Int. J. Quant. Chem. 2000, 80, 227.
- Eisenstein, O.; Lefour, J.M.; Anh, N.T.; Hudson, R.F. Tetrahedron 1977, 33, 523.
- K.N. Houk, K.N.; Li, Y.; Evanseck., J.T. Angew. Chem. Int. Ed. Engl. 1992, 31, 682. [CrossRef]
- Chandra, A. K.; Nguyen, M. T. J. Chem. Soc. Perkin Trans. 2 1997, 1415.
- Raspoet, G.; Nguyen, M. T.; McGarraghy, M.; Hegarty, A. F. J. Org. Chem. 1998, 63, 6867.Nguyen, M. T.; Raspoet, G. Can. J. Chem. 1999, 77, 817.Nguyen, M. T.; Raspoet, G.; Vanquickenborne, L. G. J. Phys. Org. Chem. 2000, 13, 46.
- Chandra, A. K.; Geerlings, P.; Nguyen, M. T. J. Org. Chem. 1997, 62, 6417.Nguyen, L. T.; Le, T. N.; De Proft, F.; Chandra, A. K.; Langenaeker, W.; Nguyen, M. T.; Geerlings, P. J. Am. Chem. Soc. 1999, 121, 5992.
- Chandra, A. K.; Nguyen, M. T. J. Comput. Chem. 1998, 19, 195.Nguyen, L. T.; Le, T. N.; De Proft, F.; Chandra, A. K.; Uchimaru, T.; Nguyen, M. T.; Geerlings, P. J. Org. Chem. 2001, 66, 6096.Le, T. N.; Nguyen, L. T.; Chandra, A. K.; De Proft, F.; Uchimaru, T.; Geerlings, P.; Nguyen, M. T. J. Chem. Soc. Perkin Trans. 2 1999, 1249.Chandra, A. K.; Uchimaru, T.; Nguyen, M. T. J. Chem. Soc. Perkin Trans. 2 1999, 2117.Chandra, A. K.; Nguyen, M. T. J. Phys. Chem. A 1998, 102, 6181.
- Sengupta, D.; Chandra, A. K.; Nguyen, M. T. J. Org. Chem. 1997, 62, 6404. [CrossRef]
- Ponti, A. J. Phys. Chem. A 2000, 104, 8843. [CrossRef]
- Rouvray, D.H. Top. Curr. Chem. 1995, 173, 2.
- Mishra, P.C.; Kumar, A. Theor. Comput. Chem. 1996, 3, 257.
- Carbo, M.; Arnau, M.; Leyda, L. Int. J. Quant. Chem. 1980, 17, 1185. [CrossRef]
- Parr, R.G.; Bartolotti, L.J. J. Phys. Chem. 1983, 87, 2810. [CrossRef]
- Boon, G.; De Proft, F.; Langenaeker, W.; Geerlings, P. Chem. Phys. Lett. 1996, 295, 122.Boon, G.; De Proft, F.; Langenaeker, W.; De Proft, F.; De Winter, H.; Tollenaere, J.P.; Geerlings, P. J. Phys. Chem.A 2001, 105, 8805.
- Spatola, A.F. Chemistry and Biochemistry of Amino Acids, Peptides and Proteins; vol. 7, Weinstein, B., Ed.; Marcel Dekker: New York, 1983; p. 267. [Google Scholar]
- Perdew, J.P.; Wang, Y. Phys. Rev. B 1992, 45, 13244. [CrossRef]
- Allmendiger, T.; Felder, E.; Hungerbühler, A. Tetr. Lett. 1995, 7301.Bartlett, P.A.; Otake, A. J. Org. Chem. 1995, 60, 3107.
- Hodgkin, E.E.; Richards, W. G. Int. J. Q. Chem., Quantum Biology Symp. 1987, 14, 1051.
- Breck, D.W. Zeolite Molecular Sieves : Structure, Chemistry and Use; John Wiley: Canada, 1974. [Google Scholar]
- Uytterhoeven, J.B.; Christner, L.G.; Hall, W.K. J. Phys. Chem. 1965, 69, 2117. [CrossRef]
- Van Bekkum, H.; Flanigen, E.M.; Jansen, J.C. Introduction to Zeolite Science and Practice; Elsevier: Amsterdam, 1991. [Google Scholar]
- Sherman, J.D. Proc. Natl. Acad. Sci. USA 1999, 96, 3471. [CrossRef]
- Van Genechten, K.; Mortier, W.J.; Geerlings, P. J. Chem. Phys. 1987, 86, 5063.
- Langenaeker, W.; De Proft, F.; Geerlings, P. Recent Developments in Physical Chemistry; Vol. 2, Transworld Research Network: Triviandum, India, 1998; p. 1219. [Google Scholar]
- Peirs, J.C.; De Proft, F.; Baron, G.; Van Alsenoy, C.; Geerlings, P. Chem. Comm. 1997, 531.
- Tielens, F.; Langenaeker, W.; Ocakoglu, A. R.; Geerlings, P. J. Comp. Chem. 2000, 21, 909.
- Tielens, F.; Geerlings, P. J. Mol. Catal. 2001, A166, 175. [CrossRef]
- Ruthven, D. M. Principles of Adsorption and Adsorption Processes; John Wiley: Canada, 1984. [Google Scholar]
- Kiselev, A. V. Pure Appl. Chem. 1980, 52, 2161.
- Tielens, F.; Geerlings, P. Int. J. Quant. Chem. 2002, 84, 58.Tielens, F.; Geerlings, P. Chem. Phys. Lett. 2002, 354, 474.
- Buckingham, A.D. Intermolecular Interactions; Pullman, B., Ed.; Wiley, 1988. [Google Scholar]
- Dunning, T.H., Jr. J. Chem. Phys. 1989, 90, 1007.
- Kendall, R.A.; Dunning Jr., T.H.; Harrison, R.J. J. Chem. Phys. 1992, 96, 6796.
- De Proft, F.; Tielens, F.; Geerlings, P. J. Mol. Struct. (Theochem) 2000, 506, 1. [CrossRef]
- Langenaeker, W.; De Proft, F.; Tielens, F.; Geerlings, P. Chem. Phys. Lett. 1998, 288, 628.
- Chatterjee, A.; Iwasaki, T.; Ebina, T. J. Phys. Chem. A 1999, 103, 2489. [CrossRef]
- Chatterjee, A.; Iwasaki, T. J. Phys. Chem. A 1999, 103, 9857. [CrossRef]
- Chatterjee, A.; Iwasaki, T. J. Phys. Chem. A 2001, 105, 6187. [CrossRef]
- Baeten, A.; Maes, D.; Geerlings, P. J. Theoret. Biol. 1998, 195, 27. [CrossRef]
- Mignon, P.; Loverix, S.; Van Houtven, S.; Steyaert, J.; Geerlings, P. in preparation.
- Carter, P.; Wells, J.A. Nature 1988, 332, 565.
- Russell, A.J.; Fersht, A.R. J. Mol. Biol. 1987, 193, 803. [CrossRef]
- Bott, R.; Vetsch, M.; Kossiakoff, A.; Graycar, T.; Katz, B.; Power, S. J. Biol. Chem. 1988, 263, 7895.
- Fersht, A. Enzyme Structure and Mechanism; W.H. Freeman and Co.: New York, 1985. [Google Scholar]
- Wells, J.A.; Estell, D.A. TIBS 1988, 13, 291, (1988).
- Geerlings, P.; De Proft, F.; Langenaeker, W. (Eds.) Density Functional Theory: a Bridge between Chemistry and Physics. In Proceedings of a Two Day Symposium at the VUB, VUB-Press, Brussels, May 14-15, 1998; 1999.
- *The subscripts (-) and (+) stand for softness vs. electrophilic and nucleophilic attacks respectively.
© 2002 by MDPI (http://www.mdpi.org), Basel, Switzerland.
Share and Cite
Geerlings, P.; De Proft, F. Chemical Reactivity as Described by Quantum Chemical Methods. Int. J. Mol. Sci. 2002, 3, 276-309. https://doi.org/10.3390/i3040276
Geerlings P, De Proft F. Chemical Reactivity as Described by Quantum Chemical Methods. International Journal of Molecular Sciences. 2002; 3(4):276-309. https://doi.org/10.3390/i3040276
Chicago/Turabian StyleGeerlings, P., and F. De Proft. 2002. "Chemical Reactivity as Described by Quantum Chemical Methods" International Journal of Molecular Sciences 3, no. 4: 276-309. https://doi.org/10.3390/i3040276
APA StyleGeerlings, P., & De Proft, F. (2002). Chemical Reactivity as Described by Quantum Chemical Methods. International Journal of Molecular Sciences, 3(4), 276-309. https://doi.org/10.3390/i3040276