Density Functional Studies of the Dipole Polarizabilities of Substituted Stilbene, Azoarene and Related Push-Pull Molecules

{kind=link}
{kind=link}
{kind=link}
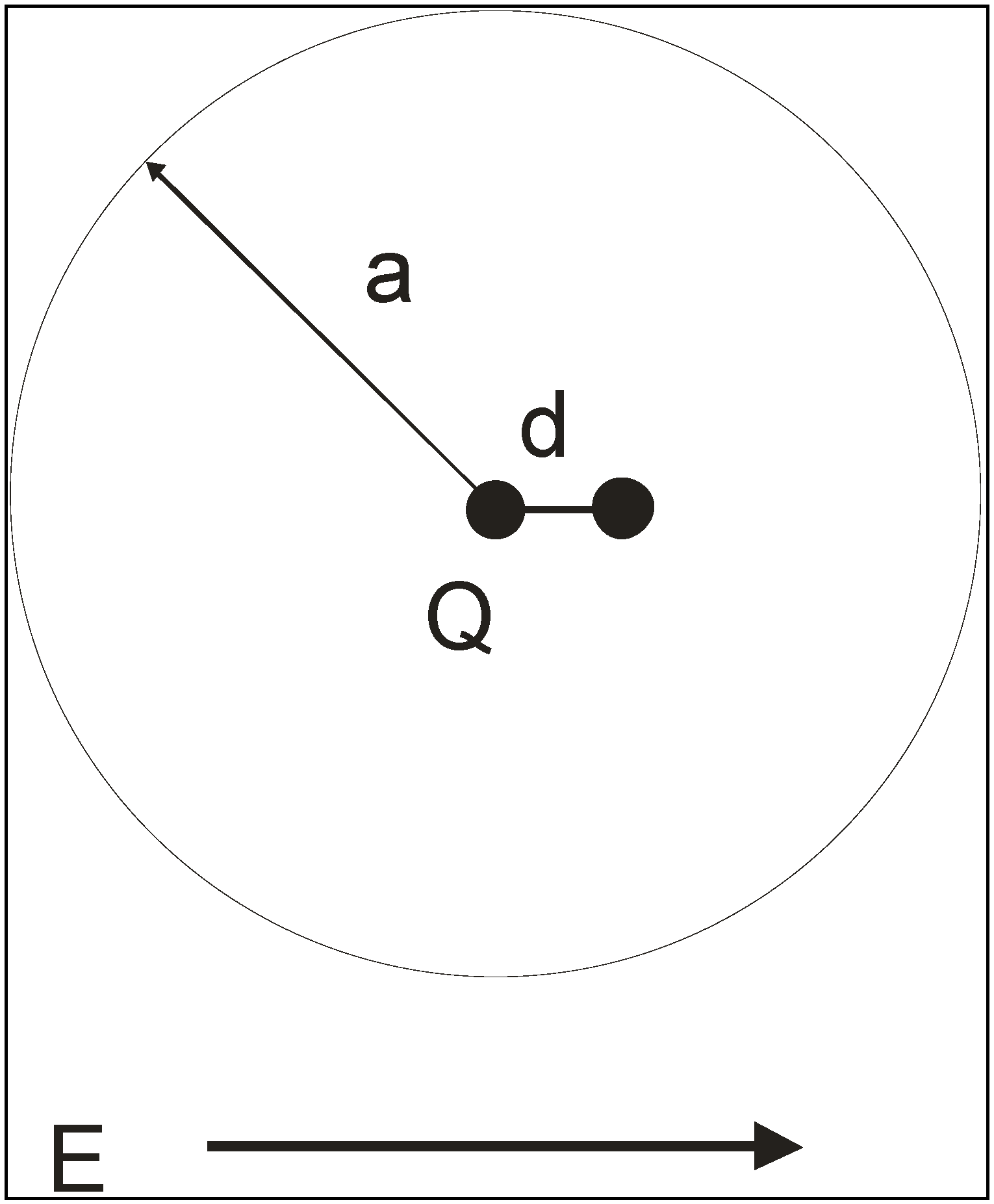
Abstract
Share and Cite
Hinchliffe, A.; Nikolaidi, B.; Soscún Machado, H.J. Density Functional Studies of the Dipole Polarizabilities of Substituted Stilbene, Azoarene and Related Push-Pull Molecules. Int. J. Mol. Sci. 2004, 5, 224-238. https://doi.org/10.3390/i5050224
Hinchliffe A, Nikolaidi B, Soscún Machado HJ. Density Functional Studies of the Dipole Polarizabilities of Substituted Stilbene, Azoarene and Related Push-Pull Molecules. International Journal of Molecular Sciences. 2004; 5(8):224-238. https://doi.org/10.3390/i5050224
Chicago/Turabian StyleHinchliffe, Alan, Beatrice Nikolaidi, and Humberto J. Soscún Machado. 2004. "Density Functional Studies of the Dipole Polarizabilities of Substituted Stilbene, Azoarene and Related Push-Pull Molecules" International Journal of Molecular Sciences 5, no. 8: 224-238. https://doi.org/10.3390/i5050224
APA StyleHinchliffe, A., Nikolaidi, B., & Soscún Machado, H. J. (2004). Density Functional Studies of the Dipole Polarizabilities of Substituted Stilbene, Azoarene and Related Push-Pull Molecules. International Journal of Molecular Sciences, 5(8), 224-238. https://doi.org/10.3390/i5050224