How the Substituent Effect Influences π-Electron Delocalisation in the Ring of Reactants in the Reaction Defining the Hammett Substituent Constants σm and σp

Abstract
:Introduction
Methods
Results and Discussion

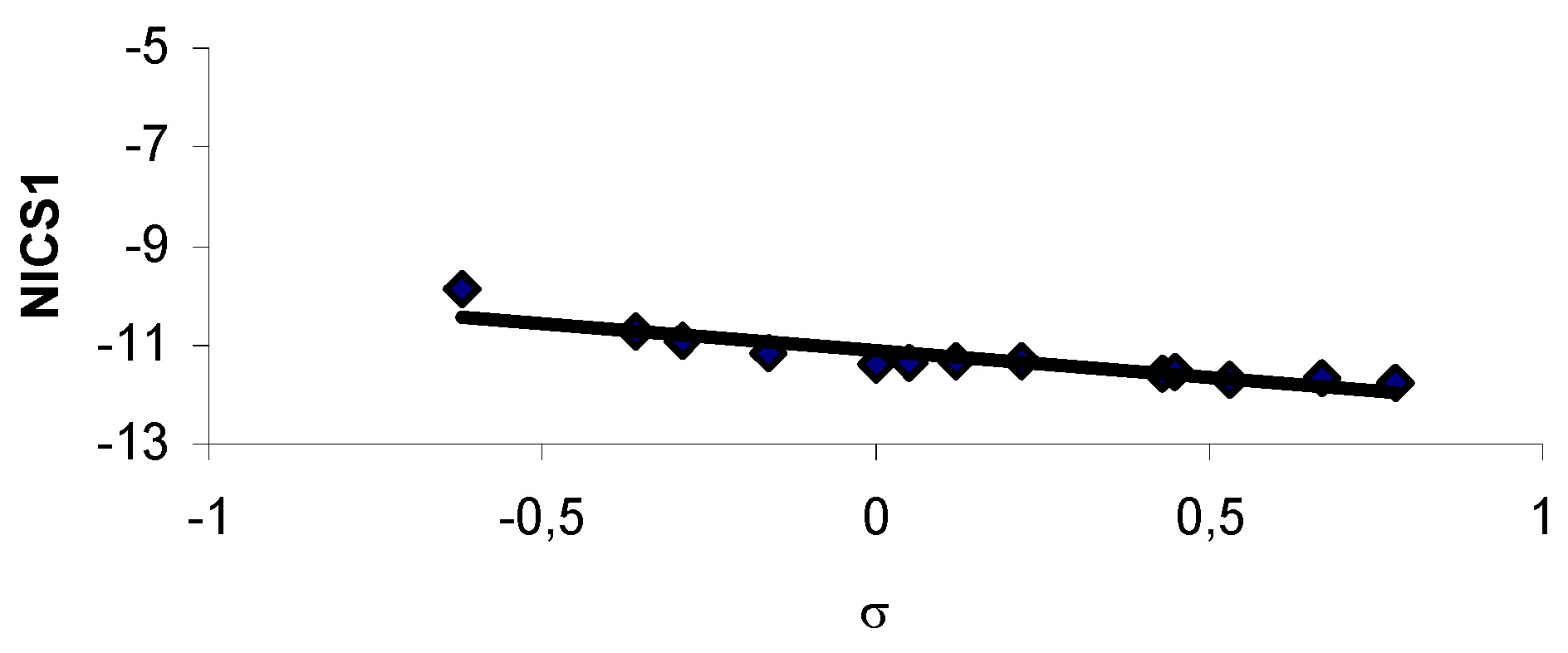{kind=link}
| Entry | Substituent | HOMA | GEO | EN | NICS | NICS1 | σ |
|---|---|---|---|---|---|---|---|
| 1 | H | 0.982 | 0.004 | 0.015 | -9.73 | -11.38 | 0 |
| 2 | 3-CH3 | 0.978 | 0.005 | 0.018 | -9.89 | -11.36 | -0.06 |
| 3 | 3-CH3Cl | 0.981 | 0.003 | 0.016 | -10.17 | -11.38 | -0.09 |
| 4 | 3-CF3 | 0.985 | 0.003 | 0.012 | -10.39 | -11.54 | 0.44 |
| 5 | 3-CHO | 0.977 | 0.005 | 0.019 | -9.64 | -11.37 | 0.36 |
| 6 | 3-COOCH3 | 0.980 | 0.003 | 0.017 | -9.75 | -11.35 | 0.33 |
| 7 | 3-CN | 0.976 | 0.005 | 0.019 | -10.38 | -11.54 | 0.62 |
| 8 | 3-NH2 | 0.973 | 0.006 | 0.021 | -10.17 | -10.70 | 0 |
| 9 | 3-NO2 | 0.988 | 0.005 | 0.007 | -10.95 | -11.58 | 0.73 |
| 10 | 3-OH | 0.984 | 0.003 | 0.013 | -11.10 | -11.32 | 0.1 |
| 11 | 3-OCH3 | 0.976 | 0.008 | 0.017 | -11.05 | -11.46 | 0.11 |
| 12 | 3-F | 0.985 | 0.010 | 0.005 | -11.85 | -11.73 | 0.34 |
| 13 | 3-Cl | 0.987 | 0.005 | 0.009 | -10.75 | -11.48 | 0.37 |
| 14 | 4-CH3 | 0.970 | 0.008 | 0.022 | -9.62 | -11.16 | -0.16 |
| 15 | 4-CH2Cl | 0.977 | 0.006 | 0.017 | -10.14 | -11.31 | 0.12 |
| 16 | 4-CF3 | 0.985 | 0.004 | 0.012 | -10.59 | -11.72 | 0.53 |
| 17 | 4-CHO | 0.972 | 0.011 | 0.018 | -9.98 | -11.57 | 0.43 |
| 18 | 4-COOCH3 | 0.977 | 0.007 | 0.016 | -10.06 | -11.54 | 0.45 |
| 19 | 4-CN | 0.972 | 0.011 | 0.018 | -10.56 | -11.66 | 0.67 |
| 20 | 4-NH2 | 0.951 | 0.025 | 0.024 | -9.08 | -9.88 | -0.62 |
| 21 | 4-NO2 | 0.987 | 0.005 | 0.007 | -11.23 | -11.76 | 0.78 |
| 22 | 4-OH | 0.977 | 0.010 | 0.013 | -10.27 | -10.74 | -0.36 |
| 23 | 4-OCH3 | 0.967 | 0.015 | 0.018 | -10.30 | -10.93 | -0.29 |
| 24 | 4-F | 0.987 | 0.008 | 0.005 | -11.33 | -11.36 | 0.05 |
| 25 | 4-Cl | 0.987 | 0.004 | 0.010 | -10.50 | -11.32 | 0.22 |
| esd | 0.0084 | 0.0049 | 0.0052 | 0.6335 | 0.4054 | 0.3548 | |
| mean | 0.978 | 0.007 | 0.015 | -10.377 | -11.325 | 0.2028 |
| Entry | Substituent | HOMA | GEO | EN | NICS | NICS1 | σ |
|---|---|---|---|---|---|---|---|
| 1 | H | 0.978 | 0.001 | 0.021 | -9.72 | -11.50 | 0 |
| 2 | 3-CH3 | 0.976 | 0.001 | 0.022 | -9.789 | -11.37 | -0.06 |
| 3 | 3-CH3Cl | 0.975 | 0.002 | 0.022 | -10.07 | -11.74 | -0.09 |
| 4 | 3-CF3 | 0.979 | 0.002 | 0.019 | -10.36 | -11.67 | 0.44 |
| 5 | 3-CHO | 0.964 | 0.009 | 0.028 | -9.42 | -11.39 | 0.36 |
| 6 | 3-COOCH3 | 0.970 | 0.005 | 0.025 | -9.62 | -11.38 | 0.33 |
| 7 | 3-CN | 0.967 | 0.002 | 0.026 | -10.34 | -11.65 | 0.62 |
| 8 | 3-NH2 | 0.977 | 0.002 | 0.022 | -10.13 | -10.79 | 0 |
| 9 | 3-NO2 | 0.985 | 0.002 | 0.013 | -10.73 | -11.57 | 0.73 |
| 10 | 3-OH | 0.985 | 0.002 | 0.014 | -11.05 | -11.36 | 0.1 |
| 11 | 3-OCH3 | 0.983 | 0.002 | 0.016 | -11.05 | -11.55 | 0.11 |
| 12 | 3-F | 0.980 | 0.013 | 0.007 | -11.85 | -11.78 | 0.34 |
| 13 | 3-Cl | 0.986 | 0.004 | 0.010 | -10.90 | -11.59 | 0.37 |
| 14 | 4-CH3 | 0.977 | 0.002 | 0.022 | -9.71 | -11.31 | -0.16 |
| 15 | 4-CH2Cl | 0.972 | 0.005 | 0.023 | -9.99 | -11.38 | 0.12 |
| 16 | 4-CF3 | 0.980 | 0.003 | 0.017 | -10.35 | -11.69 | 0.53 |
| 17 | 4-CHO | 0.956 | 0.014 | 0.029 | -9.39 | -11.40 | 0.43 |
| 18 | 4-COOCH3 | 0.963 | 0.010 | 0.027 | -9.55 | -11.36 | 0.45 |
| 19 | 4-CN | 0.960 | 0.013 | 0.028 | -10.29 | -11.63 | 0.67 |
| 20 | 4-NH2 | 0.978 | 0.001 | 0.021 | -9.95 | -10.67 | -0.62 |
| 21 | 4-NO2 | 0.978 | 0.006 | 0.015 | -10.51 | -11.48 | 0.78 |
| 22 | 4-OH | 0.984 | 0.001 | 0.015 | -10.83 | -11.23 | -0.36 |
| 23 | 4-OCH3 | 0.978 | 0.003 | 0.019 | -10.88 | -11.45 | -0.29 |
| 24 | 4-F | 0.985 | 0.008 | 0.006 | -11.66 | -11.68 | 0.05 |
| 25 | 4-Cl | 0.987 | 0.003 | 0.010 | -10.78 | -11.53 | 0.22 |
| esd | 0.0085 | 0.0042 | 0.0075 | 0.6659 | 0.2609 | 0.3549 | |
| mean | 0.976 | 0.005 | 0.019 | -10.357 | -11.446 | 0.202 |

- (i)
- increased stability when compared with acylic analogues,
- (ii)
- averaged bond lengths, intermediate between the typical single and double bonds,
- (iii)
- induction of the π-electron ring current when the system is exposed to external magnetic field and
- (iv)
- the tendency of the system to retain its π-electron structure in chemical reactions – substitution is privileged over the addition reactions.
Conclusions
Acknowledgments
References and Notes
- Institute for Scientific Information, Philadelphia (1996-2004). Retrieved in February 2004.
- (a)Exner, O. Correlation Analysis in Chemistry; Plenum Press: New York, 1997; Chap. 10; p. 439. [Google Scholar]Charton, M. Progr. Phys. Org. Chem. 1981, 13, 119.Fujio, M.; McIver, R.T.; Taft, R.W. J. Am. Chem. Soc. 1981, 103, 4017–4029.Charton, M. Progr. Phys. Org. Chem. 1987, 16, 287–315.Hansch, C.; Leo, A.; Taft, R.W. Chem. Rev. 1991, 91, 165–195.Shorter, J. J. Pure Appl. Chem. 1994, 66, 2451–2468.Shorter, J. J. Pure Appl. Chem. 1997, 69, 2497–2510.
- Hammett, L. P. Physical Organic Chemistry; McGraw – Hill: New York, 1940. [Google Scholar]
- Stock, L.M.; Brown, H.C. Adv. Phys. Org. Chem. 1990, 1, 36–154.Buncel, E.; Norris, A.R.; Russel, K.E. Quart. Rev. 1968, XXII, 123–146.(c)Hoggett, J.G.; Moodie, R.B.; Penton, R.B.; Schofield, K. Nitration and Aromatic Reactivity; Cambridge University Press: Cambridge, U.K., 1971. [Google Scholar](d)Taylor, R. Electrophilic Aromatic Substitution; Wiley: New York, 1990. [Google Scholar]
- Exner, O.; Bőhm, S. J. Org. Chem. 2002, 67, 6320–6327.
- Kruszewski, J.; Krygowski, T.M. Tetrahedron Lett. 1972, 3839–3842.Krygowski, T.M. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78.
- Krygowski, T.M.; Cyrański, M. Tetrahedron 1996, 52, 1713–1722.
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, H.; Jiao, H.; Hommes, N. J. R v. E. J. Am. Chem. Soc. 1996, 118, 6317. [CrossRef]
- Schleyer, P.v.R.; Monoharar, M.; Wang, Z.; Kiran, B.; Jiao, H.; Puchta, R.; Hommess, N.J.R.v.E. Org. Lett. 2001, 3, 2465–2468.
- Hellmann, H. Einfürung in die Quantenchemie 1937. [CrossRef]Feynman, R.P. Phys. Rev. 1937, 56, 340.
- Schleyer, P.v.R. Chem. Rev. 2001, 101, 1115–1117.
- Krygowski, T.M.; Pindelska, E.; Cyrański, M.K.; Häfelinger, G. Chem. Phys. Lett. 2002, 359, 158–162.
- Fowler, P.W.; Havenith, R.W.A.; Jenneskens, L.W.; Soncini, A.; Steiner, E. Angew. Chem. 2002, 41, 1558–1560.
- Czerminski, J.; Iwasiewicz, A.; Paszek, Z.; Sikorski, A. Statistical Methods in Applied Chemistry; PWN and Elsevier: Warsaw and Amsterdam, 1990. [Google Scholar]
- Stępień, B. T.; Krygowski, T. M.; Cyrański, M. K. J. Org. Chem. 2002, 67, 5987–5992.
- Stępień, B. T.; Krygowski, T. M.; Cyrański, M. K. J. Phys. Org. Chem. 2003, 16, 426–430. [CrossRef]
- Krygowski, T.M.; Cyrański, M.K.; Czarnocki, Z.; Häfelinger, G.; Katritzky, A.R. Tetrahedron 2000, 56, 1783–1796.Cyrański, M.K.; Krygowski, T.M.; Katritzky, A.R.; Schleyer, P.v.R. J. Org. Chem. 2002, 67, 1333–1338.
- Cyranski, M.; Krygowski, T. M. J. Chem. Inf. Comput. Sci. 1996, 36, 1142–1145. [CrossRef]
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Krygowski, T.M.; Stępień, B.T.; Cyrański, M.K. How the Substituent Effect Influences π-Electron Delocalisation in the Ring of Reactants in the Reaction Defining the Hammett Substituent Constants σm and σp. Int. J. Mol. Sci. 2005, 6, 45-51. https://doi.org/10.3390/i6010045
Krygowski TM, Stępień BT, Cyrański MK. How the Substituent Effect Influences π-Electron Delocalisation in the Ring of Reactants in the Reaction Defining the Hammett Substituent Constants σm and σp. International Journal of Molecular Sciences. 2005; 6(1):45-51. https://doi.org/10.3390/i6010045
Chicago/Turabian StyleKrygowski, Tadeusz M., Beata T. Stępień, and Michał K. Cyrański. 2005. "How the Substituent Effect Influences π-Electron Delocalisation in the Ring of Reactants in the Reaction Defining the Hammett Substituent Constants σm and σp" International Journal of Molecular Sciences 6, no. 1: 45-51. https://doi.org/10.3390/i6010045
APA StyleKrygowski, T. M., Stępień, B. T., & Cyrański, M. K. (2005). How the Substituent Effect Influences π-Electron Delocalisation in the Ring of Reactants in the Reaction Defining the Hammett Substituent Constants σm and σp. International Journal of Molecular Sciences, 6(1), 45-51. https://doi.org/10.3390/i6010045