Identification and molecular modeling of a family 5 endocellulase from Thermus caldophilus GK24, a cellulolytic strain of Thermus thermophilus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing of a whole genome shot gun library of Thermus calodphilus GK24
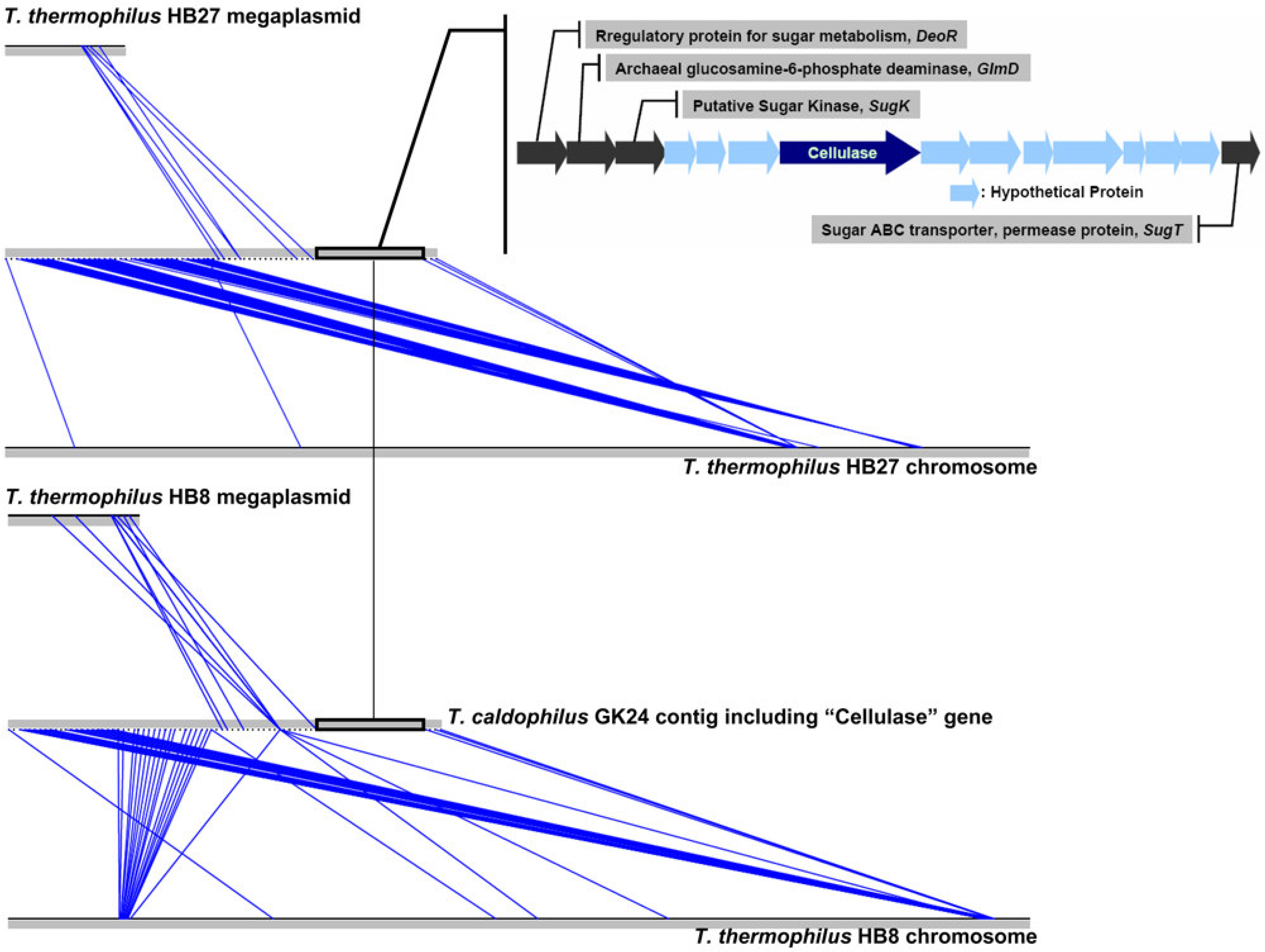
2.2. Data Processing, Sequence Assembly, and Annotation
2.3. Detection of endocellulase activity
2.4. Purification of the enzyme
2.5. Activities of enzyme against various substrates
2.6. Measurement of the optimum temperature
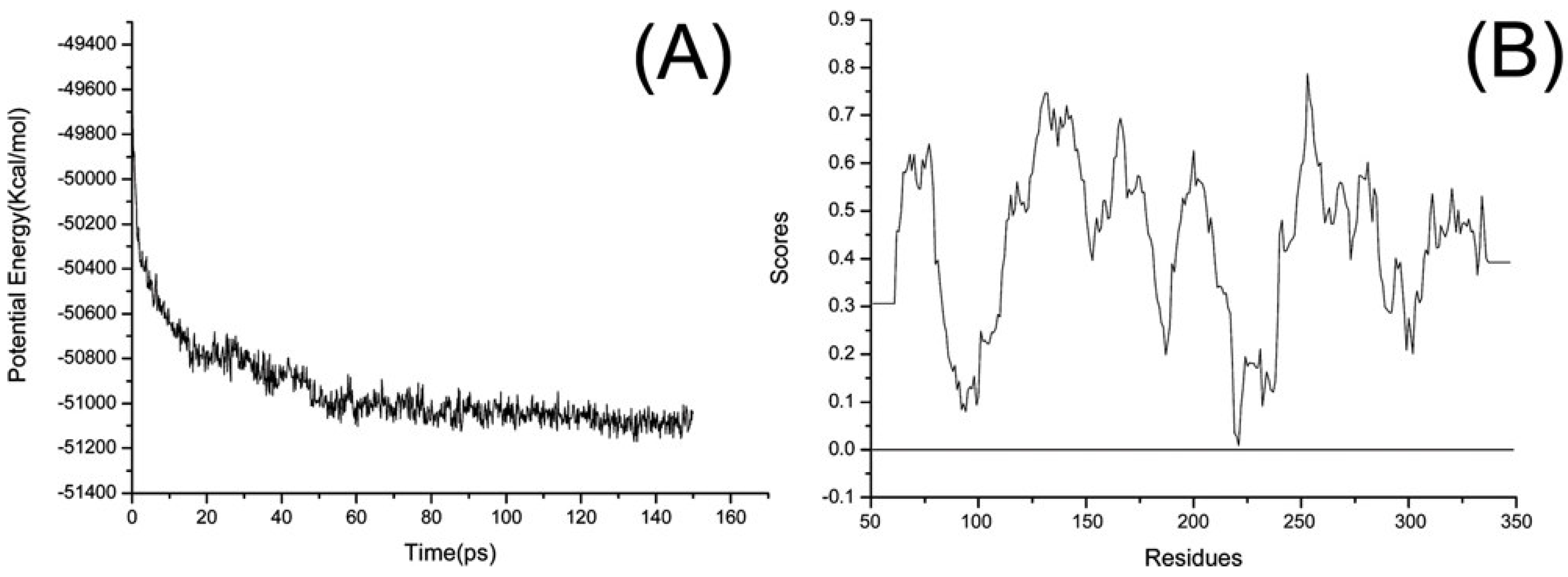
2.7. Homology modeling of T. caldophilus GK24 Endocellulase
2.8. Identification of the binding site of TcCel5A
2.9. Molecular modeling of cellotetraose docking into theTcCel5A binding site
3. Results and discussion
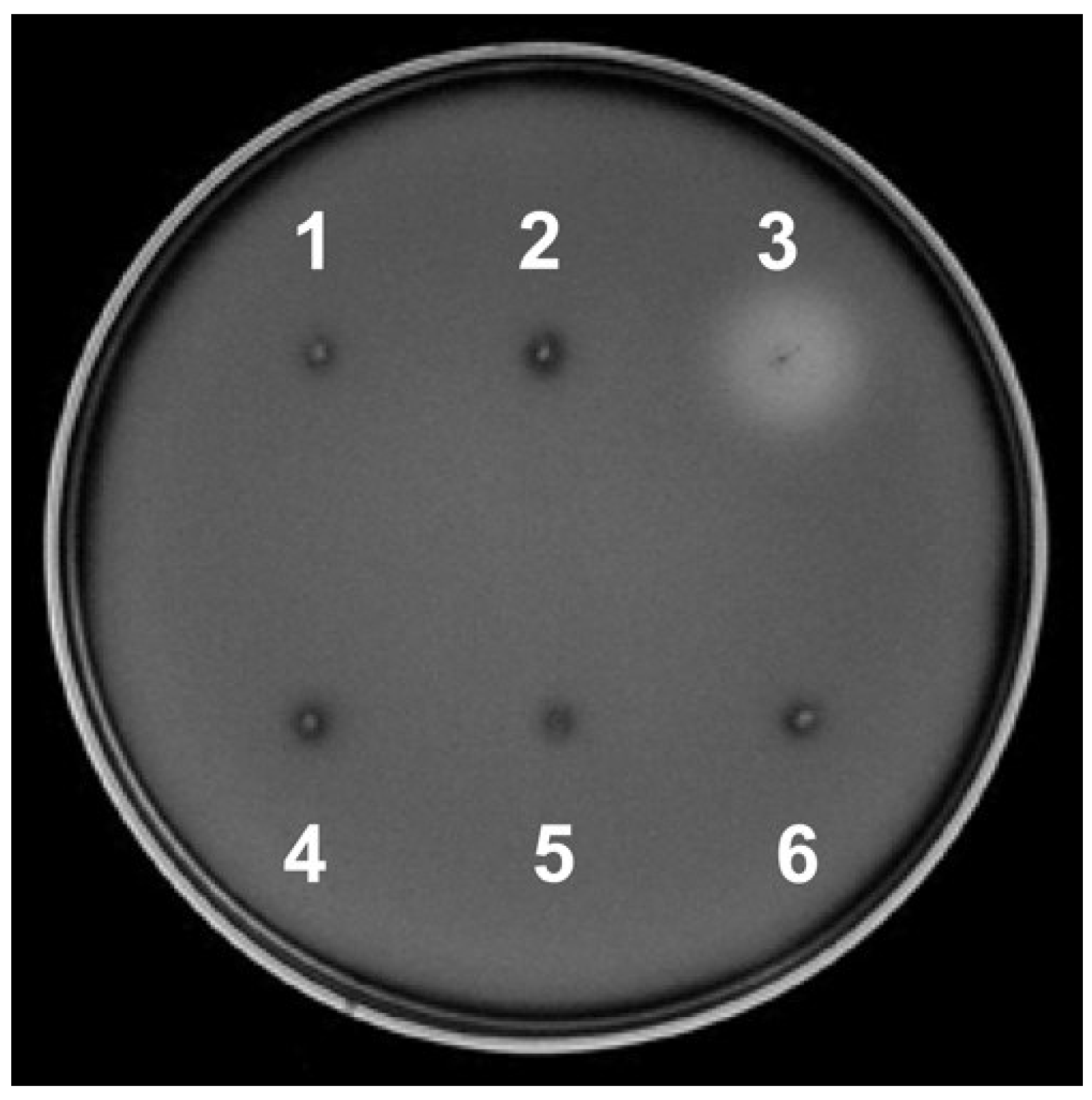
3.1. Endocellulase activity of T. caldophilus GK24


3.2. Purification and Activity of TcCel5A against various substrates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | TcCel5A (units/mg) |
|---|---|
| Carboxymethyl-cellulose (CMC) | 23.2 |
| Alkali swollen cellulose (ASC) | 9.16 |
| Insoluble cellooligosaccharide (ICOS) | 8.21 |
| Cellotriose | N.D.a |
| Cellotetraose | 21.9 |
| Cellopentaose | 16.5 |
| Cellohexaose | 7.84 |

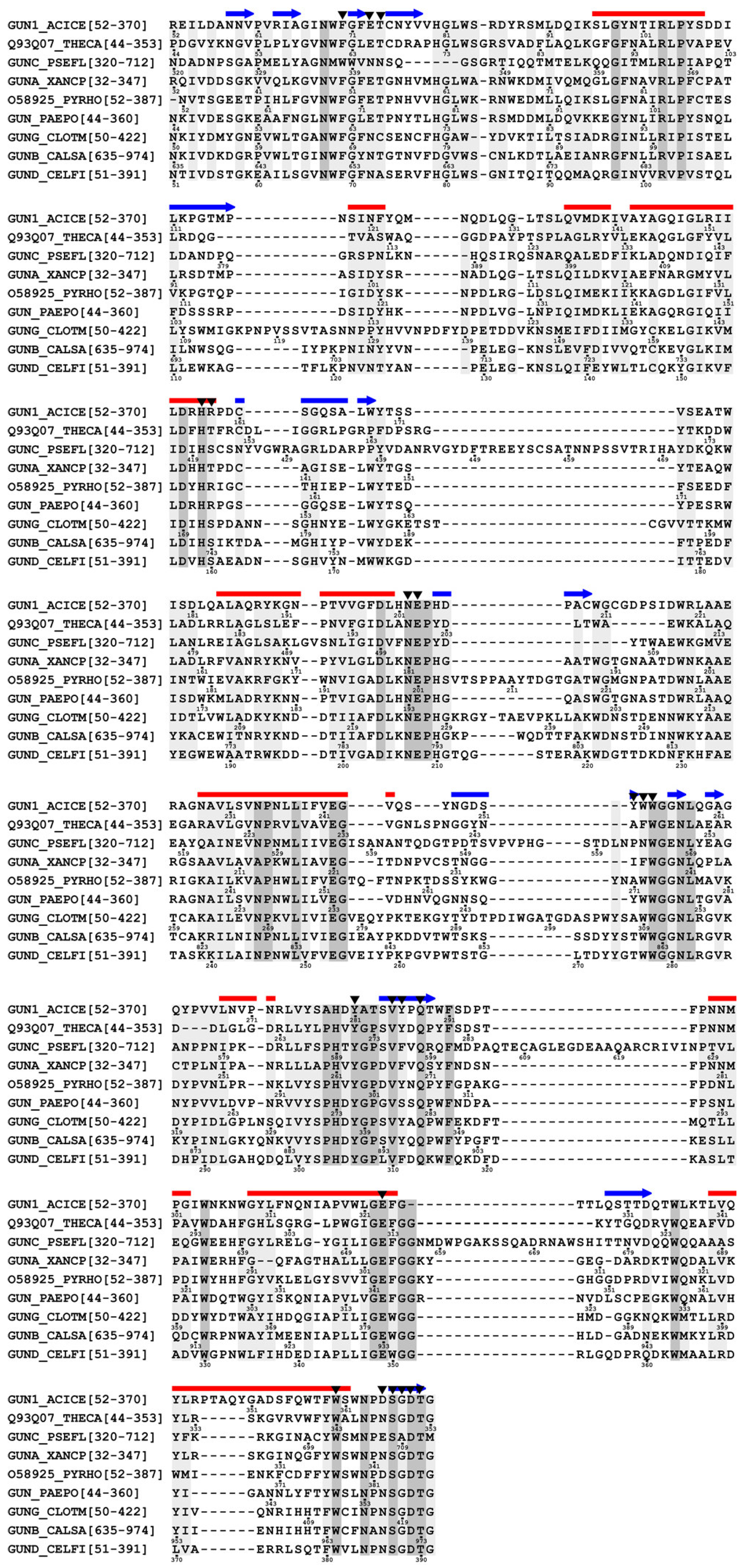
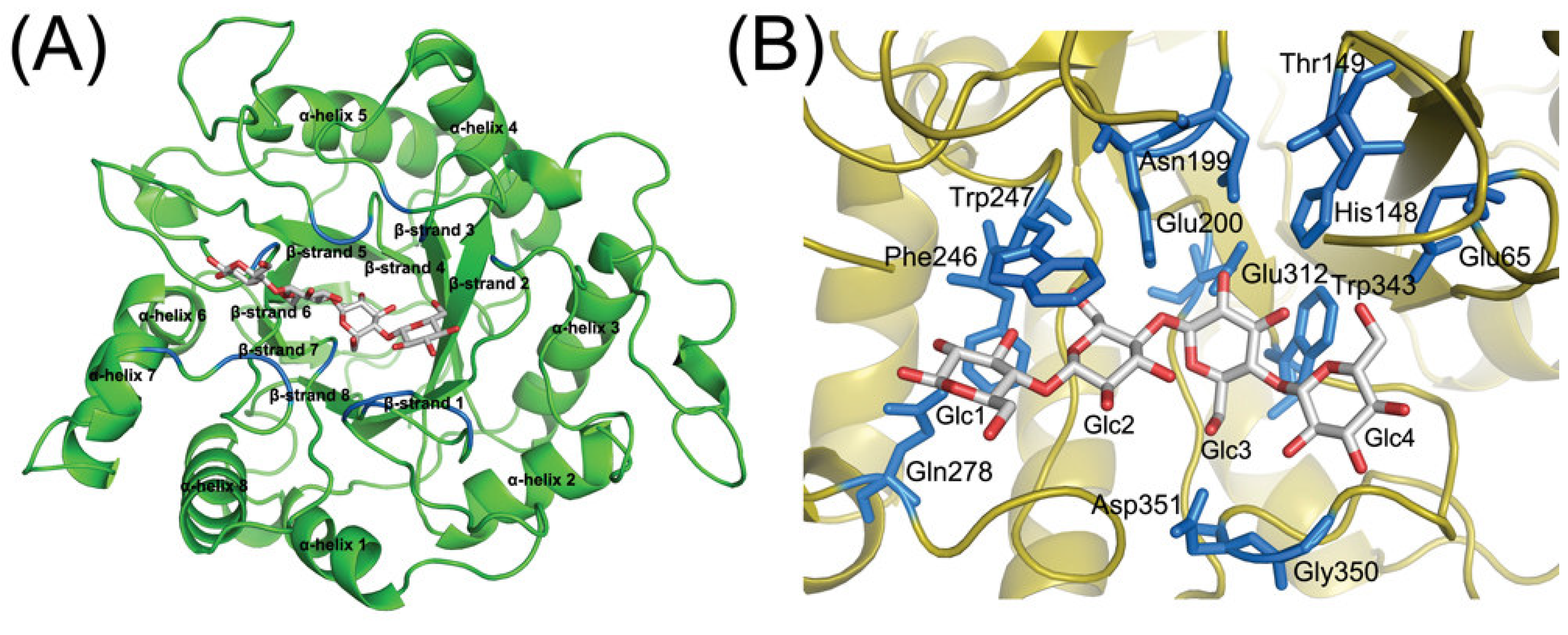
3.3. Homology modeling of TcCel5A


| Target protein | AcCel5A | TcCel5A |
|---|---|---|
| %U–W Angles in core Ramachandran region | 96.7% | 95.6% |
| Number bond distances with significant deviations | 0 | 0 |
| Number bond angles with significant deviations | 2 | 8 |
| Number residues examined | 317 | 308 |

3.4. Identification of the binding site of TcCel5A
3.5. Explicit characterization of the TcCel5A-G4 complex
| Cellotetraose | Atom | Classa | Residue | Atom | Classa | Distb | Surfc |
|---|---|---|---|---|---|---|---|
| Glc1 | O1 | I | Tyr276 | OH | I | 4.8 | 0.2 |
| O1 | I | Gln278 | OE1 | II | 4.9 | 1.4 | |
| O2 | I | Gln278 | NE2 | III | 3.0 | 16.5 | |
| O2 | I | Ala245 | O | II | 3.7 | 15.4 | |
| O2 | I | Gln278 | OE1 | II | 4.1 | 0.3 | |
| O3 | I | Trp247 | N | III | 3.6 | 1.2 | |
| O5 | II | Trp247 | NE1 | III | 4.4 | 1.4 | |
| Glc2 | O2 | I | Val275 | O | II | 5.5 | 0.2 |
| O6 | I | Trp247 | N | III | 3.0 | 26.7 | |
| O6 | I | Trp247 | O | II | 3.2 | 10.4 | |
| O6 | I | Glu200 | OE2 | II | 3.4 | 0.2 | |
| Glc3 | O2 | I | Glu312 | OE1 | II | 3.0 | 10.4 |
| O2 | I | Glu312 | OE2 | II | 3.1 | 9.0 | |
| O2 | I | Glu200 | OE2 | II | 3.2 | 5.5 | |
| O2 | I | Glu200 | OE1 | II | 3.3 | 1.9 | |
| O2 | I | Asn199 | ND2 | III | 3.3 | 5.2 | |
| O3 | I | His148 | NE2 | I | 3.0 | 18.4 | |
| O4 | II | Trp343 | NE1 | III | 3.7 | 0.7 | |
| O6 | I | Asp351 | OD1 | II | 2.8 | 13.7 | |
| O6 | I | Asp351 | OD2 | II | 4.2 | 0.3 | |
| Glc4 | O2 | I | Gly350 | N | III | 3.6 | 9.4 |
| O2 | I | Asp351 | OD1 | II | 4.2 | 0.5 | |
| O3 | I | Gly350 | N | III | 3.3 | 14.7 | |
| O3 | I | Asn348 | O | II | 3.7 | 7.4 | |
| O4 | I | Thr149 | OB | III | 4.5 | 2.6 | |
| O4 | I | Asn348 | O | II | 4.7 | 0.7 | |
| O6 | I | Glu65 | OE2 | II | 2.7 | 13.7 | |
| O6 | I | Thr149 | OB | III | 3.4 | 8.1 | |
| O6 | I | His148 | NE2 | I | 3.6 | 7.1 |
| Residue | Dista | Surfb | HBc | ARd | PHe | DCf | Evdw | Eele | Etotal |
|---|---|---|---|---|---|---|---|---|---|
| Glu312* | 3.0 | 27.9 | + | - | - | - | 2.199 | -37.633 | -35.434 |
| Glu200* | 2.5 | 39.5 | + | - | - | + | 1.007 | -14.473 | -13.466 |
| Asn199* | 3.3 | 5.2 | + | - | - | - | -5.176 | 0.385 | -4.791 |
| Asp351* | 2.7 | 31.6 | + | - | - | - | -3.398 | -1.268 | -4.666 |
| His148* | 2.8 | 31.1 | + | - | - | - | -3.496 | -0.757 | -4.253 |
| Trp343* | 3.0 | 26.4 | + | - | - | - | -2.619 | -0.845 | -3.464 |
| Trp247* | 3.0 | 18.7 | + | - | - | - | -2.667 | -0.432 | -3.099 |
| Glu65* | 3.0 | 108.1 | + | - | - | + | -2.702 | -0.166 | -2.868 |
| Gly350* | 3.3 | 28.9 | + | - | - | - | -1.324 | -1.428 | -2.752 |
| Gln278* | 3.5 | 26.0 | + | - | - | - | -1.297 | -1.434 | -2.731 |
| Asn348 | 3.7 | 8.1 | + | - | - | - | -2.931 | 0.268 | -2.663 |
| Thr149* | 3.4 | 29.4 | + | - | - | - | -1.108 | -1.435 | -2.543 |
| Phe246* | 3.2 | 37.3 | - | - | - | + | -2.332 | -0.166 | -2.498 |
| Ala245 | 3.7 | 15.9 | + | - | - | - | -2.447 | 0.106 | -2.341 |
| Ser349* | 3.7 | 15.9 | - | - | - | - | -2.080 | -0.257 | -2.337 |
| Phe62* | 3.7 | 30.4 | - | - | - | - | -2.184 | -0.127 | -2.311 |
| Tyr271* | 3.7 | 36.5 | - | - | - | - | -1.796 | -0.513 | -2.309 |
| Tyr276* | 3.9 | 49.5 | + | - | - | + | -4.953 | 2.942 | -2.011 |
| Val275* | 4.1 | 22.4 | + | - | - | + | -1.790 | 0.135 | -1.655 |
| Thr352* | 4.3 | 17.3 | - | - | - | - | -1.690 | 0.059 | -1.631 |
| Thr66* | 4.3 | 8.8 | - | - | - | + | -1.414 | -0.052 | -1.466 |
| His269* | 4.7 | 0.9 | - | - | - | + | 0.516 | -1.858 | -1.342 |
| Total | -43.682 | -58.949 | -102.631 |
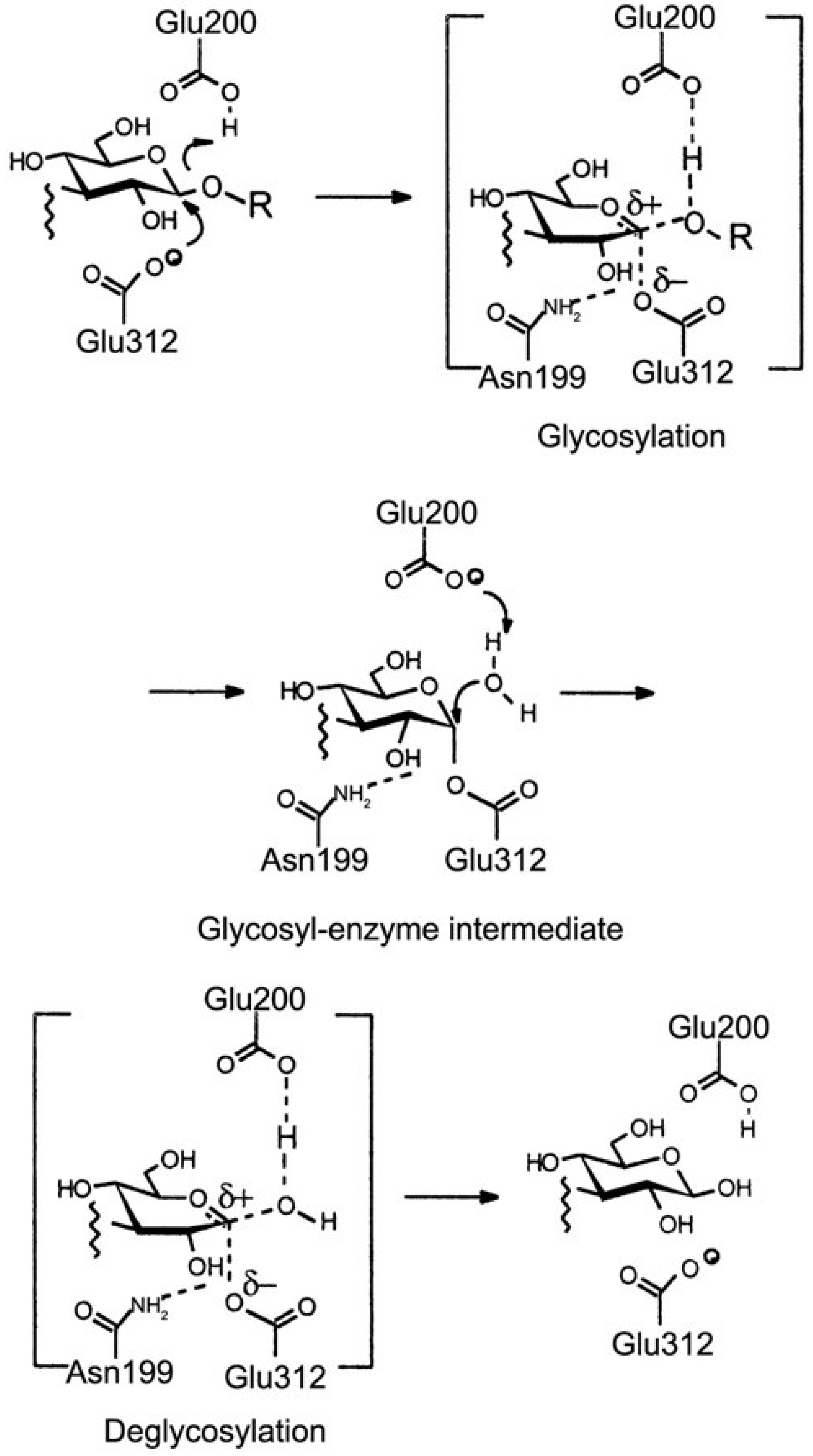
3.6. Proposed mechanism of hydrolysis by Thermus caldophilus GK24 endocellulase

4. Conclusions
Acknowledgements
References and Notes
- da Costa, M.S.; Nobre, M.F.; Rainey, R.A. The genus Thermus. In Bergey’s manual of systematic bacteriology, 2nd ed.; vol. 1, Boone, D.R., Castenholtz, R.W., Eds.; Springer: New York, N.Y., 2001. [Google Scholar]
- Weisburg, W.G.; Giovannoni, S.J.; Woese, C.R. The Deinococcus-Thermus phylum and the effect of rRNA composition on phylogenetic tree construction. Syst. Appl. Micorbiol. 1989, 11, 128–134. [Google Scholar] [CrossRef]
- Brock, T.D.; Freeze, H. Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J. Bacterial. 1969, 98, 289–297. [Google Scholar]
- Taguchi, H.; Yamashita, M.; Matsuzawa, H.; Ohta, T. Heat-stable and fructose 1,6-bisphosphate-activated L-lactate dehydrogenase from an extremely thermophilic bacterium. J. Biochem. (Tokyo) 1982, 91, 1343–1348. [Google Scholar]
- Oshima, T.; Imahori, K. Isolation of an extreme thermophile and thermostabilities of its transfer ribonucleic acid and ribosomes. J. Gen. Appl. Microbiol. 1971, 17, 513–317. [Google Scholar] [CrossRef]
- Manaia, C.M.; Hoste, B.; Gutierrez, M.C.; Gillis, M.; Ventosa, A.; Kersters, K.; da Costa, M.S. Halotolerant Thermus strains from marine and terrestrial hot springs belong to Thermus thermophilus nom. rev. emend. Syst. Appl. Microbiol. 1994, 17, 526–532. [Google Scholar]
- Williams, R.A.; Smith, K.E.; Welch, S.G.; Micallef, J.; Sharp, R.J. DNA relatedness of Thermus strains, description of Thermus brockianus sp. nov., and proposal to reestablish Thermus thermophilus. Int. J. Syst. Bacteriol. 1995, 45, 495–499. [Google Scholar] [CrossRef]
- Nobre, M.F.; Truper, H.G.; da Costa, M.S. Transfer of Thermus ruber, Thermus silvanus and Thermus chliarophilus to Meiothermus gen. nov. as Meiothermus ruber comb. nov., Meiothermus silvanus comb. nov. and Meiothermus chliarophlus comb. nov., respectively, and emendation of the genus Thermus. Int. J. Syst. Bacterial. 1996, 46, 604–606. [Google Scholar] [CrossRef]
- Coughlan, M.P. Cellulose degradation by fungi, in Microbial Enzymes and Biotechnology; Fogarty, W.M., Kelly, C.T., Eds.; Elsevier Applied Science: London, 1990. [Google Scholar]
- Ito, S. Alkaline cellulases from alkaliphilic Bacillus: enzymatic properties, genetics, and application to detergents. Extremophiles 1997, 1, 61–66. [Google Scholar] [CrossRef]
- Beguin, P.; Aubert, J.A. The biological degradation of cellulose. FEMS Microbiol. Rev. 1994, 13, 25–58. [Google Scholar] [CrossRef]
- Tomme, P.; Warren, R.A.J.; Gilkes, N.R. Cellulose Hydrolysis by Bacteria and Fungi. Adv. Microbial Physiol. 1985, 37, 1–81. [Google Scholar] [CrossRef]
- Gilkes, N.R.; Kilburn, D.G.; Miller, R.C., Jr.; Warren, R.A.J. Structural and functional analysis of a bacterial cellulase by proteolysis. J. Biol. Chem. 1989, 264, 17802–17808. [Google Scholar]
- Gilkes, N.R.; Henrissat, B.; Kilburn, D.G.; Miller, R.C., Jr.; Warren, R.A.J. Domains in microbial β-1,4-glycanase: Sequence conservation, function, and enzyme families. Microbiol. Rev. 1991, 55, 303–315. [Google Scholar]
- Stone, J.E.; Scallan, A.M.; Donefer, E.; Ahlgren, E. Cellulases and their application; Gould, R.F., Ed.; American Chemical Society: Washington DC, 1969. [Google Scholar]
- Fan, L.T.; Lee, Y.H.; Beardmore, D.H. Mechanism of the enzymatic hydrolysis of cellulose: Effects of major structural features of cellulose on enzymatic hydrolysis. Biotechnol. Bioeng. 1980, 22, 177–199. [Google Scholar]
- Cowling, E.B. Physical and chemical constraints in the hydrolysis of cellulose and lignocellulosic materials. Biotechno. Bioeng. Symp. 1975, 5, 163–181. [Google Scholar]
- Grethlein, H.E. The effect of pore size distribution on the rate of enzymatic hydrolysis of cellulosic substrates. Bio/Technology 1985, 3, 155–160. [Google Scholar] [CrossRef]
- Gama, F.M.; Teixeira, J.A.; Mota, M. Cellulose morphology and enzymatic reactivity: A modified solute exclusion technique. Biotechnol. Bioeng. 1994, 43, 381–387. [Google Scholar] [CrossRef]
- Lee, S.B.; Shin, H.S.; Ryu, D.D.Y. Adsorption of cellulase on cellulose: Effect of physicochemical properties of cellulose on adsorption and rate of hydrolysis. Biotechnol. Bioeng. 1982, 24, 2137–2153. [Google Scholar] [CrossRef]
- Woodward, J.; Hayes, M.K.; Lee, N.E. Hydrolysis of cellulose by saturating and non-saturating concentrations of cellulase: Implications for synergism. Bio/Technology 1988, 6, 301–304. [Google Scholar] [CrossRef]
- Tanaka, M.; Ikesaka, M.; Matsuno, R. Effect of Pore Size in Substrate and Diffusion of Enzyme on Hydrolysis of Cellulosic Materials with Cellulases. Biotechnol. Bioeng. 1988, 32, 698–706. [Google Scholar] [CrossRef]
- Coughlan, M.P. Mechanisms of cellulose degradation by fungi and bacteria. Anim. Feed Sci. Tech. 1991, 32, 77–100. [Google Scholar] [CrossRef]
- Enari, T.M.; Niku-Paavola, M.L. Enzymatic hydrolysis of cellulose: is the current theory of the mechanisms of hydrolysis valid? CRC Cri. Rev. Biotech. 1987, 5, 67–87. [Google Scholar] [CrossRef]
- Wood, T.M.; Garcia-Campayo, V. Enzymology of cellulose degradation. Biodegradation 1990, 1, 147. [Google Scholar] [CrossRef]
- Woodward, J. Synergism in cellulase systems. Bioresource Tech. 1991, 36, 67–75. [Google Scholar] [CrossRef]
- Henrissat, B. Enzymatic cellulose degradation. Cellulose Commun. 1998, 5, 84–90. [Google Scholar]
- Sinnot, M.L. Catalytic mechanisms of enzymic glycosyl transfer. Chem. Rev. 1990, 90, 1171–1202. [Google Scholar] [CrossRef]
- Rabinovich, M.L.; Melnick, M.S.; Bolobova, A.V. The structure and mechanism of action of cellulolytic enzymes. Biochemistry (Moscow) 2002, 67, 850–871. [Google Scholar] [CrossRef]
- Skopec, C.E.; Himmel, M.E.; Matthews, J.F.; Brady, J.W. Energetics for displacing a single chain from the surface of microcrystalline cellulose into the active site of Acidothermus cellulolyticus Cel5A. Protein Eng. 2003, 16, 1005–1015. [Google Scholar] [CrossRef]
- McCarter, S.L.; Adney, W.S.; Vinzant, T.B.; Jennings, E.; Eddy, F.P.; Decker, S.R.; Baker, J.O.; Himmel, M.E. Exploration of cellulose surface-binding properties of Acidothermus cellulolyticus Cel5A by site-specific mutagenesis. Appl. Biochem. Biotech. - Part A Enzyme Engineering and Biotechnology 2002, 98–100, 273–287. [Google Scholar] [CrossRef]
- Sakon, J.; Adney, W.S.; Himmel, M.E.; Thomas, S.R.; Karplus, P.A. Crystal structure of thermostable family 5 endocellulase E1 from Acidothermus cellulolyticus in complex with cellotetraose. Biochemistry 1996, 35, 10648–10660. [Google Scholar] [CrossRef]
- Zverlov, V.V.; Schantz, N.; Schwarz, W.H. A major new component in the cellulosome of Clostridium thermocellum is a processive endo-β-1,4-glucanase producing cellotetraose. FEMS Microbiol. Let. 2005, 249, 353–358. [Google Scholar] [CrossRef]
- Guimarães, B.G.; Souchon, H.; Lytle, B.L.; Wu, J.H.D.; Alzari, P.M. The crystal structure and catalytic mechanism of cellobiohydrolase celS, the major enzymatic component of the Clostridium thermocellum cellulosome. J. Mol. Biol. 2002, 320, 587–596. [Google Scholar] [CrossRef]
- Zang, S.; Guifang, L.; David, B.W. Characterization of a Thermomonospora fusca Exocellulase. Biochemistry 1995, 34, 3386–3395. [Google Scholar] [CrossRef]
- Harchand, R.K.; Singh, S. Characterization of cellulase complex of Streptomyces albaduncus. J. Basic Microbiol. 1997, 37, 93–103. [Google Scholar] [CrossRef]
- Jin-Duck, B.; Dinesh, A.Y.; Douglas, E.E. Purification, Characterization, and Molecular Analysis of Thermostable cellulases CelA and CelB from Thermotoga neapolitana. Appl. Environ. Microbiol. 1998, 64, 4774–4781. [Google Scholar]
- Tettelin, H.; Radune, D.; Kasif, S.; Khouri, H.; Salzberg, S.L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics 1999, 62, 500–507. [Google Scholar] [CrossRef]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Gordon, D.; Abajian, C.; Green, P. Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8, 195–202. [Google Scholar] [CrossRef]
- Gordon, D.; Desmarais, C.; Green, P. Automated finishing with autofinish. Genome Res. 2001, 11, 614–625. [Google Scholar] [CrossRef]
- Park, J.H.; Park, B.C.; Koh, S.H.; Kim, J.S.; Koh, J.H.; Yang, M.H.; Kim, Y.S.; Kim, C.H.; Kim, M.H.; Kwon, S.T.; Lee, D.S. Genome mapping of an extreme thermophile, Thermus caldophilus GK24. Genomics inform. 2003, 1, 50–54. [Google Scholar]
- Henne, A.; Bruggemann, H.; Raasch, C.; Wiezer, A.; Hartsch, T.; Liesegang, H.; Johann, A.; Lienard, T.; Gohl, O.; Martinez-Arias, R.; Jacobi, C.; Starkuviene, V.; Schlenczeck, S.; Dencker, S.; Huber, R.; Klenk, H.P.; Kramer, W.; Merkl, R.; Gottschalk, G.; Fritz, H.J. The genome sequence of the extreme thermophile Thermus thermophilus. Nat. Biotechnol. 2004, 22, 547–553. [Google Scholar] [CrossRef]
- Available online: http://gib.genes.nig.ac.jp/single/main.php?spid=Tthe_HB8.
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Teather, R.M.; Wood, P.J. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl. Environ. Microbiol. 1982, 43, 777–780. [Google Scholar]
- Tsigelny, I.F. Protein Structural Prediction Bioinformatic approach, International University Line. 2002.
- Pearson, W.R. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics 1991, 11, 635–650. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Sali, A.; Overington, J.P. Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci. 1994, 3, 1582–1596. [Google Scholar] [CrossRef]
- Sali, A.; Potterton, L.; Yuan, F.; Van Vlijmen, H.; Karplus, M. Evaluation of comparative protein modeling by MODELLER. Proteins 1995, 23, 318–326. [Google Scholar] [CrossRef]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assesment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Discover 3 User Guide; San Diego: MSI, USA, 1999.
- Miller, G.L. Method in carbohydrate chemistry; Whistler, R.L., Ed.; Academic Press: New York, 1963; Vol III, pp. 134–139. [Google Scholar]
- Ponder, J.W.; Richards, F.M. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 1987, 193, 775–791. [Google Scholar] [CrossRef]
- Tull, D.; Withers, S.G.; Gilkes, N.R.; Kilburn, D.G.; Warren, R.A.J.; Aebersold, R. Glutamic acid 274 is the nucleophile in the active site of a "retaining" exoglucanase from Cellulomonas fimi. J. Biol. Chem. 1991, 266, 15621–15625. [Google Scholar]
- Wang, Q.; Tull, D.; Meinke, A.; Gilkes, N.R.; Warren, R.A.J.; Aebersold, R.; Withers, S.G. Glu280 is the nucleophile in the active site of Clostridium thermocellum CelC, a family A endo-beta-1,4-glucanase. J. Biol. Chem. 1993, 268, 14096–14102. [Google Scholar]
- Bortoli-German, I.; Haiech, J.; Chippaux, M.; Barras, F. Informational Suppression to Investigate Structural Functional and Evolutionary Aspects of the Erwinia chrysanthemi Cellulase. J. Mol. Biol. 1995, 246, 82–94. [Google Scholar] [CrossRef]
- Planas, A. Bacterial 1,3-1,4-β-glucanases: Structure, function and protein engineering. Biochim. Biophys. Acta. 2000, 1543, 361–382. [Google Scholar]
- Jenkins, J.; Leggio, L.L.; Harris, G.; Pickersgill, R. Beta-glucosidase, beta-galactosidase, family A cellulases, family F xylanases and two barley glycanases form a superfamily of enzymes with 8-fold beta/alpha architecture and with two conserved glutamates near the carboxy-terminal ends of beta-strands four and seven. FEBS Lett. 1995, 362, 281–285. [Google Scholar] [CrossRef] [Green Version]
© 2006 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, D.; Park, B.H.; Jung, B.-W.; Kim, M.-k.; Hong, S.-I.; Lee, D.-S. Identification and molecular modeling of a family 5 endocellulase from Thermus caldophilus GK24, a cellulolytic strain of Thermus thermophilus. Int. J. Mol. Sci. 2006, 7, 571-589. https://doi.org/10.3390/i7120571
Kim D, Park BH, Jung B-W, Kim M-k, Hong S-I, Lee D-S. Identification and molecular modeling of a family 5 endocellulase from Thermus caldophilus GK24, a cellulolytic strain of Thermus thermophilus. International Journal of Molecular Sciences. 2006; 7(12):571-589. https://doi.org/10.3390/i7120571
Chicago/Turabian StyleKim, Dooil, Bo Hyun Park, Bo-Won Jung, Mi-kyung Kim, Suk-In Hong, and Dae-Sil Lee. 2006. "Identification and molecular modeling of a family 5 endocellulase from Thermus caldophilus GK24, a cellulolytic strain of Thermus thermophilus" International Journal of Molecular Sciences 7, no. 12: 571-589. https://doi.org/10.3390/i7120571
APA StyleKim, D., Park, B. H., Jung, B.-W., Kim, M.-k., Hong, S.-I., & Lee, D.-S. (2006). Identification and molecular modeling of a family 5 endocellulase from Thermus caldophilus GK24, a cellulolytic strain of Thermus thermophilus. International Journal of Molecular Sciences, 7(12), 571-589. https://doi.org/10.3390/i7120571