Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
2.1. Computational details
2.2. Results
2.3. Discussion
2.4. Conclusion
3. Experimental Section
Inhibition of CA II
Topological indexes
Electrostatic indexes
- DPSA-1 Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC].
- RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC].
Acknowledgments
References and Notes
- Lehtonen, JM; Parkkila, S; Vullo, D; Casini, A; Scozzafava, A; Supuran, CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozyme XIII with aromatic and heterocyclic sulfonamides: a novel target for the drug design. Bioorg Med Chem Lett 2004, 14, 3757–3762. [Google Scholar]
- Supuran, CT; Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007, 15, 4336–4350. [Google Scholar]
- Carbonic Anhydrase Its Inhibitors and Activators; Supuran, CT; Scozzafava, A; Conway, J (Eds.) CRC Press: Boca Raton, FL, USA, 2004; pp. 1–363, and references cited therein.
- Parkkila, S. An Overview of the Distribution and Function of Carbonic Anhydrase in Mammals. In The Carbonic Anhydrases—New Horizons; Chegwidden, WR, Edwards, Y, Carter, N, Eds.; Birkhäuser Verlag: Basel, Switzerland, 2000; pp. 79–93. [Google Scholar]
- Karelson, M. Molecular Descriptors in QSAR/QSPR; Wiley: New York, NY, 2000. [Google Scholar]
- Tadeschini, R; Consonni, V. Handbook of Molecular Descriptors; Wiley–VCH: Weinheim, Germany, 2000. [Google Scholar]
- Clare, BW; Supuran, CT. Carbonic anhydrase inhibitors. Part 86. A QSAR study on some sulfonamide drugs which lower intra-ocular pressure, using the ACE non-linear statistical method. Eur J Med Chem 2000, 35, 859–865. [Google Scholar]
- Clare, BW; Supuran, CT. Carbonic anhydrase inhibitors. Part 61. Quantum chemical QSAR of a group of benzenedisulfonamides. Eur J Med Chem 1999, 34, 463–474. [Google Scholar]
- Supuran, CT; Clare, BW. Carbonic anhydrase inhibitors – Part 57: Quantum chemical QSAR of a group of 1,3,4-thiadiazole- and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999, 34, 41–50. [Google Scholar]
- Supuran, CT; Clare, BW. Carbonic anhydrase inhibitors-Part 47: Quantum chemical quantitative structure-activity relationships for a group of sulfanilamide Schiff base inhibitors of carbonic Anhydrase. Eur J Med Chem 1998, 33, 489–500. [Google Scholar]
- Clare, BW; Supuran, CT. Carbonic anhydrase inhibitors. Part 41. Quantitative structure-activity correlations involving kinetic rate constants of 20 sulfonamide inhibitors from a non-congeneric series. Eur J Med Chem 1997, 32, 311–319. [Google Scholar]
- Supuran, CT; Clare, BW. Carbonic anhydrase inhibitors. Part 24. A quantitative structure-activity relationship study of positively charged sulfonamide inhibitors. Eur J Med Chem 1995, 30, 687–696. [Google Scholar]
- Eroglu, E; Türkmen, H. A DFT-based quantum theoretic QSAR study of aromatic and heterocyclic sulfonamides as carbonic anhydrase inhibitors against isozyme, CA-II. Journal of Molecular Graphics and Modelling 2007, 26, 701–708. [Google Scholar]
- Eroglu, E; Türkmen, H; Güler, S; Palaz, S; Oltulu, O. A DFT-Based QSARs study of Acetazolamide/Sulfanilamide derivatives with carbonic anhydrase (CA-II) isozyme inhibitory activity. Int J Mol Sci 2007, 8, 145–155. [Google Scholar]
- Agrawal, VK; Singh, J; Khadikar, PV; Supuran, CT. QSAR study on topically acting sulfonamides incorporating GABA moieties: A molecular connectivity approach. Bioorg Med Chem Lett 2006, 16, 2044–2051. [Google Scholar]
- Melagraki, G; Afantitis, A; Sarimveis, H; Iglessi-Markopoulou, O; Supuran, CT. QSAR study on para-substituted aromatic sulfonamides as carbonic anhydrase II inhibitors using topological information indices. Bioorg Med Chem 2006, 14, 1108–1114. [Google Scholar]
- Agrawal, VK; Banerji, M; Gupta, M; Singh, J; Khadikar, PV; Supuran, CT. QSAR study on carbonic anhydrase inhibitors: water-soluble sulfonamides incorporating β-alanyl moieties, possessing long lasting-intra ocular pressure lowering properties-a molecular connectivity approach. Eur J Med Chem 2005, 40, 1002–1012. [Google Scholar]
- Khadikar, PV; Sharma, V; Karmarkar, S; Supuran, CT. QSAR studies on benzene sulfonamide carbonic anhydrase inhibitors: need of hydrophobic parameter for topological modeling of binding constants of sulfonamides to human CA-II. Bioorg Med Chem Lett 2005, 15, 923–930. [Google Scholar]
- Jaiswal, M; Khadikar, PV; Supuran, CT. Topological modeling of lipophilicity, diuretic activity, and carbonic inhibition activity of benzene sulfonamides: a molecular connectivity approach. Bioorg Med Chem Lett 2004, 14, 5661–5666. [Google Scholar]
- Agrawal, VK; Bano, S; Supuran, CT; Khadikar, PV. QSAR study on carbonic anhydrase inhibitors: aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties, with topical activity as antiglaucoma agents. Eur J Med Chem 2004, 39, 593–600. [Google Scholar]
- Jaiswal, M; Khadikar, PV; Scozzafava, A; Supuran, CT. Carbonic anhydrase inhibitors: the first QSAR study on inhibition of tumor-associated isoenzyme IX with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2004, 14, 3283–3290. [Google Scholar]
- Thakur, A; Thakur, M; Khadikar, PV; Supuran, CT; Sudele, P. QSAR study on benzenesulphonamide carbonic anhydrase inhibitors: topological approach using Balaban index. Bioorg Med Chem 2004, 12, 789–793. [Google Scholar]
- Agrawal, VK; Sharma, R; Khadikar, PV. QSAR Studies on Carbonic Anhydrase Inhibitors: A Case of Ureido and Thioureido Derivatives of Aromatic/Heterocyclic Sulfonamides. Bioorg Med Chem 2002, 10, 2993–2999. [Google Scholar]
- Khadikar, PV; Clare, BW; Balaban, AT; Supuran, CT; Agrawal, VK; Singh, J; Joshi, AK; Lakwani, M. QSAR modeling of carbonic anhydrase-I, -II and -IV inhibitory activities: Relative correlation potential of six topological indices. Revue Roumaine De Chimie 2006, 51, 703–717. [Google Scholar]
- Huang, H; Pan, X; Tan, N; Zeng, N; Ji, C. 3D-QSAR study of sulfonamide inhibitors of humancarbonic anhydrase II. Eur J Med Chem 2007, 42, 365–372. [Google Scholar]
- Agrawal, VK; Singh, J; Pandey, A; Khadikar, PV. Modelling of inhibitory activities of sulphanilamide Schiff bases using physicochemical properties. Oxidation Communications 2006, 29, 803–816. [Google Scholar]
- Supuran, CT; Clare, BW. Orbital symmetry in QSAR: Some Schiff's base inhibitors of carbonic anhydrase. SAR and QSAR in Environmental Research 2001, 12, 17–29. [Google Scholar]
- Agrawal, VK; Srivastava, S; Khadikar, PV; Supuran, CT. Quantitative Structure–Activity Relationship Study on Sulfanilamide Schiff's Bases: Carbonic Anhydrase (CA) Inhibitors. Bioorg Med Chem 2003, 11, 5353–5362. [Google Scholar]
- CODESSA (Comprehensive Descriptors for Structural and Statistical Analysis), Semichem, 7204, Mullen, Shawnee, KS 66216 USA, Copyright© Semichem and the University of Florida, 1995–2004.
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA, Jr; Vreven, T; Kudin, KN; Burant, JC; Millam, JM; Iyengar, SS; Tomasi, J; Barone, V; Mennucci, B; Cossi, M; Scalmani, G; Rega, N; Petersson, GA; Nakatsuji, H; Hada, M; Ehara, M; Toyota, K; Fukuda, R; Hasegawa, J; Ishida, M; Nakajima, T; Honda, Y; Kitao, O; Nakai, H; Klene, M; Li, X; Knox, JE; Hratchian, HP; Cross, JB; Adamo, C; Jaramillo, J; Gomperts, R; Stratmann, RE; Yazyev, O; Austin, AJ; Cammi, R; Pomelli, C; Ochterski, JW; Ayala, PY; Morokuma, K; Voth, GA; Salvador, P; Dannenberg, JJ; Zakrzewski, VG; Dapprich, S; Daniels, AD; Strain, MC; Farkas, O; Malick, DK; Rabuck, AD; Raghavachari, K; Foresman, JB; Ortiz, JV; Cui, Q; Baboul, AG; Clifford, S; Cioslowski, J; Stefanov, BB; Liu, G; Liashenko, A; Piskorz, P; Komaromi, I; Martin, RL; Fox, DJ; Keith, T; Al-Laham, MA; Peng, CY; Nanayakkara, A; Challacombe, M; Gill, PMW; Johnson, B; Chen, W; Wong, MW; Gonzalez, C; Pople, JA. Gaussian 03, Revision B05; Gaussian, Inc: Pittsburgh, PA, 2003. [Google Scholar]
- Parr, RG; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, 1989. [Google Scholar]
- Becke, AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993, 98, 5648–5648. [Google Scholar]
- CODESSATM, References Manual, V. 2.13 (PC)., Semichem, 7204, Mullen, Shawnee, KS, USA, Copyright Semichem and the University of Florida, 2002.
- Drug Discov Today. 2005, 10, pp. 1497–500. (a) http://www.vcclab.org/lab/alogPs/start.html. (b) Tetko, I. V. Computing chemistry on the web.
- Supuran, CT; Scozzafava, A; Popescu, A; Bobes-Tureac, R; Banciu, A; Bobes-Tureac, G; Bamciu, MD. Carbonic anhydrase inhibitors. Part 43. Schiff bases derived from aromatic sulfonamides: towards more specific inhibitors for membrane-bound versus cytosolic isozymes. Eur J Med Chem 1997, 32, 445–452. [Google Scholar]
- Krungkrai, J; Scozzafava, A; Reungprapavut, R; Krungkrai, SR; Rattanajak, R; Kamchonwongpaisand, S; Supuran, CT. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: towards antimalarials with a novel mechanism of action. Bioorg Med Chem 2005, 13, 483–489. [Google Scholar]
- Puccetti, L; Fasolis, G; Vullo, D; Chohan, ZH; Scozzafava, A; Supuran, CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, IX, and XII with Schiff's bases incorporating chromone and aromatic sulfonamide moieties, and their zinc complexes. Bioorg Med Chem Lett 2005, 15, 3096–3101. [Google Scholar]
- Mayers, RH. Classical and Modern Regression with Applications; PWSKENT Publ. Co: Boston, MA, 1990. [Google Scholar]
- Computational Chemical Graph Theory; Basak, S; Niemi, GJ; Veith, GD; Rouvray, DH (Eds.) Nova Science Publishers: New York, NY, 1990.
- Randic, M. Characterization of molecular branching. J Am Chem Soc 1975, 97, 6609–6615. [Google Scholar]
- Kier, LB; Hall, LH. Molecular Connectivity in Chemistry and Drug Reasearch; Academic Press: New York, NY, 1976. [Google Scholar]
- Kier, L. B. Use Of Molecular Negentropy To Encode Structure Governing Biological-Activity. J Pharm Sci 1980, 69, 807–810. [Google Scholar]
- Bonchev, D. Information Theoretic Indices for Characterization of Chemical Structure; Wiley-Interscience: New York, NY, 1983. [Google Scholar]
- Basak, SC; Harriss, DK; Magnuson, VR. Comparative-Study Of Lipophilicity Versus Topological Molecular Descriptors In Biological Correlations. J Pharm Sci 1984, 73, 429–437. [Google Scholar]
- Stanton, DT; Jurs, PC. Development And Use Of Charged Partial Surface-Area Structural Descriptors In Computer-Assisted Quantitative Structure Property Relationship Studies. Anal Chem 1990, 62, 2323–2329. [Google Scholar]
- Stanton, DT; Egolf, LM; Jurs, PC; Hicks, MG. Computer-Assisted Prediction Of Normal Boiling Points Of Pyrans and Pyrroles. J Chem Inf Comput Sci 1992, 32, 306–316. [Google Scholar]
- Zefirov, NS; Kirpichenok, MA; Ismailov, FF; Trofimov, MI. Calculation schemes for atomic electronegativities in molecular graphs within the framework of sanderson principle. Dokl Akad Nauk SSSR 1987, 296, 883–887. [Google Scholar]
- Kirpichenok, MA; Zefirov, NS. Electronegativity and geometry of molecules. 1. Principles of developed approach and analysis of the effect of nearest electrostatic interactions on the bond length in organic molecules. Zh Org Khim 1987, 23, 673–691. [Google Scholar]



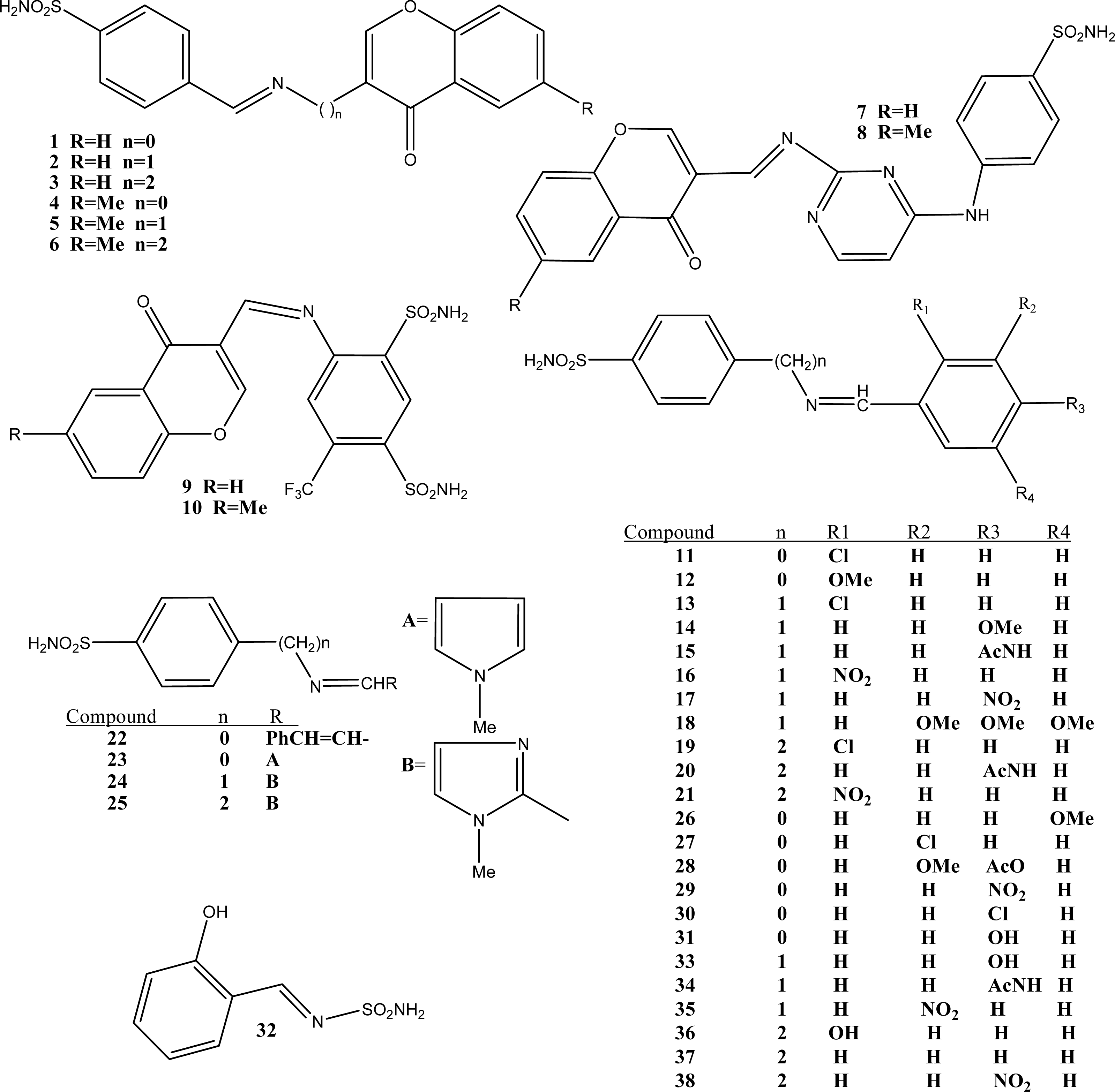{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | RNBR | NBR | NCA | RNDB | DPSA-1 | MiPCN | ALS | 3χ | RPCG | NNA | ALP | 2AIC | Obs. Ki | Pre. Ki | Residue |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.0571 | 2.0000 | 16.0000 | 0.1351 | 21.4428 | -0.0912 | -4.0600 | 8.3122 | 0.0934 | 2.0000 | 1.4900 | 4.1579 | 1.447a | 1.6557 | 0.2087 |
| 2 | 0.0526 | 2.0000 | 17.0000 | 0.1250 | 68.0099 | -0.0967 | -4.1400 | 8.5538 | 0.0894 | 2.0000 | 1.6600 | 4.3006 | 1.146a | 1.5640 | 0.418 |
| 3 | 0.0488 | 2.0000 | 18.0000 | 0.1163 | 105.7782 | -0.0980 | -4.1300 | 8.8038 | 0.0855 | 2.0000 | 1.9700 | 4.4307 | 1.322a | 1.4996 | 0.1776 |
| 4 | 0.0526 | 2.0000 | 17.0000 | 0.1250 | 54.8184 | -0.0912 | -4.3100 | 8.6547 | 0.0902 | 2.0000 | 1.9000 | 4.3951 | 1.544a | 1.7243 | 0.1803 |
| 5 | 0.0488 | 2.0000 | 18.0000 | 0.1163 | 113.8093 | -0.0967 | -4.4600 | 8.8962 | 0.0865 | 2.0000 | 2.0800 | 4.4512 | 1.740a | 1.5507 | -0.1893 |
| 6 | 0.0455 | 2.0000 | 19.0000 | 0.1087 | 165.0231 | -0.0980 | -4.3200 | 9.1462 | 0.0828 | 5.0000 | 2.3900 | 4.5694 | 1.778a | 1.4747 | -0.3033 |
| 7 | 0.0444 | 2.0000 | 20.0000 | 0.1042 | 59.8359 | -0.1043 | -4.7700 | 10.8703 | 0.0693 | 5.0000 | 1.7700 | 4.6356 | 1.518a | 1.6333 | 0.1153 |
| 8 | 0.0417 | 2.0000 | 21.0000 | 0.0980 | 117.0900 | -0.1043 | -4.9300 | 11.2127 | 0.0675 | 3.0000 | 2.1800 | 4.8192 | 1.612a | 1.6494 | 0.0374 |
| 9 | 0.0465 | 2.0000 | 17.0000 | 0.1556 | -111.7607 | -0.0872 | -5.4600 | 10.5070 | 0.1504 | 3.0000 | 2.0600 | 4.4434 | 2.008a | 1.9995 | -0.0085 |
| 10 | 0.0435 | 2.0000 | 18.0000 | 0.1458 | -67.6019 | -0.0872 | -5.9000 | 10.8494 | 0.1474 | 2.0000 | 2.4600 | 4.6297 | 2.123a | 2.0154 | -0.1076 |
| 11 | 0.0667 | 2.0000 | 13.0000 | 0.0968 | 31.6593 | -0.0932 | -4.2600 | 6.3063 | 0.1267 | 2.0000 | 2.3900 | 3.7736 | 2.461b | 2.3724 | -0.0886 |
| 12 | 0.0588 | 2.0000 | 14.0000 | 0.0857 | 130.0701 | -0.0935 | -3.7900 | 6.6081 | 0.1082 | 2.0000 | 1.9000 | 3.9476 | 2.230b | 2.1247 | -0.1053 |
| 13 | 0.0606 | 2.0000 | 14.0000 | 0.0882 | 20.6325 | -0.0986 | -4.3900 | 6.5479 | 0.1193 | 2.0000 | 2.4700 | 3.9535 | 1.954b | 2.2916 | 0.3376 |
| 14 | 0.0541 | 2.0000 | 15.0000 | 0.0789 | 138.1794 | -0.1001 | -3.9200 | 6.8399 | 0.1040 | 3.0000 | 1.9300 | 4.0539 | 2.079b | 1.9578 | -0.1212 |
| 15 | 0.0500 | 2.0000 | 16.0000 | 0.0976 | 111.5054 | -0.1002 | -3.8000 | 7.0673 | 0.0950 | 3.0000 | 1.2600 | 4.2531 | 2.041b | 1.8666 | -0.1744 |
| 16 | 0.0571 | 2.0000 | 14.0000 | 0.0833 | -18.8479 | -0.0984 | -4.0900 | 7.0830 | 0.1145 | 3.0000 | 1.8300 | 4.0436 | 2.477b | 2.3815 | -0.0955 |
| 17 | 0.0571 | 2.0000 | 14.0000 | 0.0833 | -22.6421 | -0.1000 | -4.1100 | 7.1305 | 0.1169 | 2.0000 | 1.8300 | 3.9864 | 2.146b | 2.3257 | 0.1797 |
| 18 | 0.0444 | 2.0000 | 17.0000 | 0.0652 | 291.3417 | -0.0991 | -3.9900 | 8.2996 | 0.0693 | 2.0000 | 1.6400 | 3.9908 | 1.301b | 1.2556 | -0.0454 |
| 19 | 0.0556 | 2.0000 | 15.0000 | 0.0811 | 65.0019 | -0.0999 | -4.4000 | 6.7979 | 0.1125 | 3.0000 | 2.8900 | 4.1144 | 2.322b | 2.2884 | -0.0336 |
| 20 | 0.0465 | 2.0000 | 17.0000 | 0.0909 | 140.9603 | -0.1015 | -3.9200 | 7.3173 | 0.0906 | 3.0000 | 1.6000 | 4.3855 | 1.602b | 1.8128 | 0.2108 |
| 21 | 0.0500 | 2.0000 | 15.0000 | 0.0732 | 122.8380 | -0.1001 | -4.0600 | 7.3330 | 0.1367 | 3.0000 | 1.6100 | 4.2719 | 2.397b | 2.2632 | -0.1338 |
| 22 | 0.0588 | 2.0000 | 15.0000 | 0.1143 | 40.6266 | -0.0956 | -3.7700 | 6.2710 | 0.1287 | 2.0000 | 2.5200 | 3.7556 | 1.698b | 1.8331 | 0.1351 |
| 23 | 0.0323 | 1.0000 | 12.0000 | 0.1250 | 90.8476 | -0.1147 | -2.5200 | 6.0563 | 0.1241 | 3.0000 | 0.6900 | 4.1682 | 1.000b | 1.0004 | 0.0004 |
| 24 | 0.0303 | 1.0000 | 12.0000 | 0.1176 | 155.8272 | -0.1064 | -2.1200 | 6.2979 | 0.1070 | 4.0000 | -0.3000 | 4.3549 | 1.301b | 1.0857 | -0.2153 |
| 25 | 0.0278 | 1.0000 | 13.0000 | 0.1081 | 209.1037 | -0.1064 | -2.3600 | 6.5479 | 0.1015 | 4.0000 | 0.1200 | 4.4823 | 1.000b | 1.0497 | 0.0497 |
| 26 | 0.0645 | 2.0000 | 13.0000 | 0.0938 | 43.8927 | -0.0934 | -3.2500 | 6.3063 | 0.1498 | 2.0000 | 1.5000 | 3.8574 | 2.230c | 2.2027 | -0.0273 |
| 27 | 0.0667 | 2.0000 | 13.0000 | 0.0968 | -5.1570 | -0.0932 | -4.2600 | 6.3063 | 0.1267 | 2.0000 | 2.4500 | 3.7736 | 2.462c | 2.3894 | -0.0726 |
| 28 | 0.0500 | 2.0000 | 16.0000 | 0.0976 | 186.4766 | -0.0941 | -4.1900 | 7.6016 | 0.0993 | 2.0000 | 1.5200 | 4.3929 | 2.000c | 1.8939 | -0.1061 |
| 29d | 0.0625 | 2.0000 | 13.0000 | 0.0909 | -51.7829 | -0.0946 | -4.0500 | 6.8889 | 0.1241 | 3.0000 | 1.7600 | 3.8125 | 1.698c | -- | -- |
| 30 | 0.0667 | 2.0000 | 13.0000 | 0.0968 | -23.5748 | -0.0946 | -4.2700 | 6.1901 | 0.1294 | 2.0000 | 2.4800 | 3.7069 | 2.447c | 2.3327 | -0.1143 |
| 31 | 0.0645 | 2.0000 | 13.0000 | 0.0938 | 32.0889 | -0.0947 | -3.3500 | 6.1901 | 0.1511 | 2.0000 | 1.3700 | 3.7929 | 2.278c | 2.1029 | -0.1751 |
| 32 | 0.0476 | 1.0000 | 7.0000 | 0.1429 | 2.8615 | -0.0749 | -2.2300 | 3.5542 | 0.1785 | 2.0000 | 0.6400 | 3.7257 | 1.602c | 1.7672 | 0.1652 |
| 33 | 0.0588 | 2.0000 | 14.0000 | 0.0857 | 61.6262 | -0.1001 | -3.3600 | 6.4317 | 0.1432 | 2.0000 | 1.4100 | 3.9698 | 1.845c | 2.0079 | 0.1629 |
| 34 | 0.0500 | 2.0000 | 16.0000 | 0.0976 | 112.8082 | -0.1002 | -3.8000 | 7.0673 | 0.0950 | 3.0000 | 1.2300 | 4.2531 | 2.041c | 1.8581 | -0.1829 |
| 35 | 0.0571 | 2.0000 | 14.0000 | 0.0833 | -22.7851 | -0.0997 | -4.1100 | 7.0624 | 0.1164 | 3.0000 | 1.8400 | 4.1007 | 2.255c | 2.4402 | 0.1852 |
| 36 | 0.0541 | 2.0000 | 15.0000 | 0.0789 | 135.7274 | -0.1001 | -3.3700 | 6.7979 | 0.1350 | 2.0000 | 2.0600 | 4.1824 | 2.113c | 2.1202 | 0.0072 |
| 37 | 0.0556 | 2.0000 | 15.0000 | 0.0811 | 112.8082 | -0.1017 | -3.7200 | 6.2710 | 0.1224 | 2.0000 | 2.3200 | 3.8565 | 2.146c | 1.8751 | -0.2709 |
| 38d | 0.0526 | 2.0000 | 15.0000 | 0.0769 | 8.6366 | -0.1013 | -4.1900 | 7.3805 | 0.1103 | 3.0000 | 2.2200 | 4.1427 | 1.301c | -- | -- |
| Models | Descriptors involved | t-test | CoefficientAi (i=1,2,3,4,5) | B (intercept) | Statistical Parameters N R2 R2 cv F s2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Equ. 1 | RNBR Relative number of benzene rings | 6.163 | 32.767(5.316) | 0.180 | 36 | 0.527 | 0.488 | 37.98 | 0.090 |
| (0.280) | |||||||||
| Equ. 2 | NBR Number of benzene rings | 7.590 | 1.296(0.170) | 1.245 | 36 | 0.647 | 0.599 | 30.27 | 0.069 |
| NCA Number of C atoms | -5.745 | -0.119(0.020) | (0.298) | ||||||
| Equ. 3 | RNDB Relative number of double bonds | -7.939 | -16.73(2.107) | 6.182 | 36 | 0.729 | 0.618 | 28.74 | 0.055 |
| DPSA-1 Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC] | -5.458 | -0.0031(0.0005) | (0.776) | ||||||
| MiPCN Minimum partial charge for a N atom [Zefirov's PC] | 3.406 | 24.276(7.127) | |||||||
| Equ. 4 | ALS Average logS | -5.887 | -0.462(0.078) | 1.061 | 36 | 0.786 | 0.716 | 29.98 | 0.045 |
| 3χ Randic index (order 3) | -2.571 | -0.125(0.048) | (0.330) | ||||||
| RNDB Relative number of double bonds | -4.515 | -9.122(2.020) | |||||||
| RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC] | 3.924 | 7.577(1.930) | |||||||
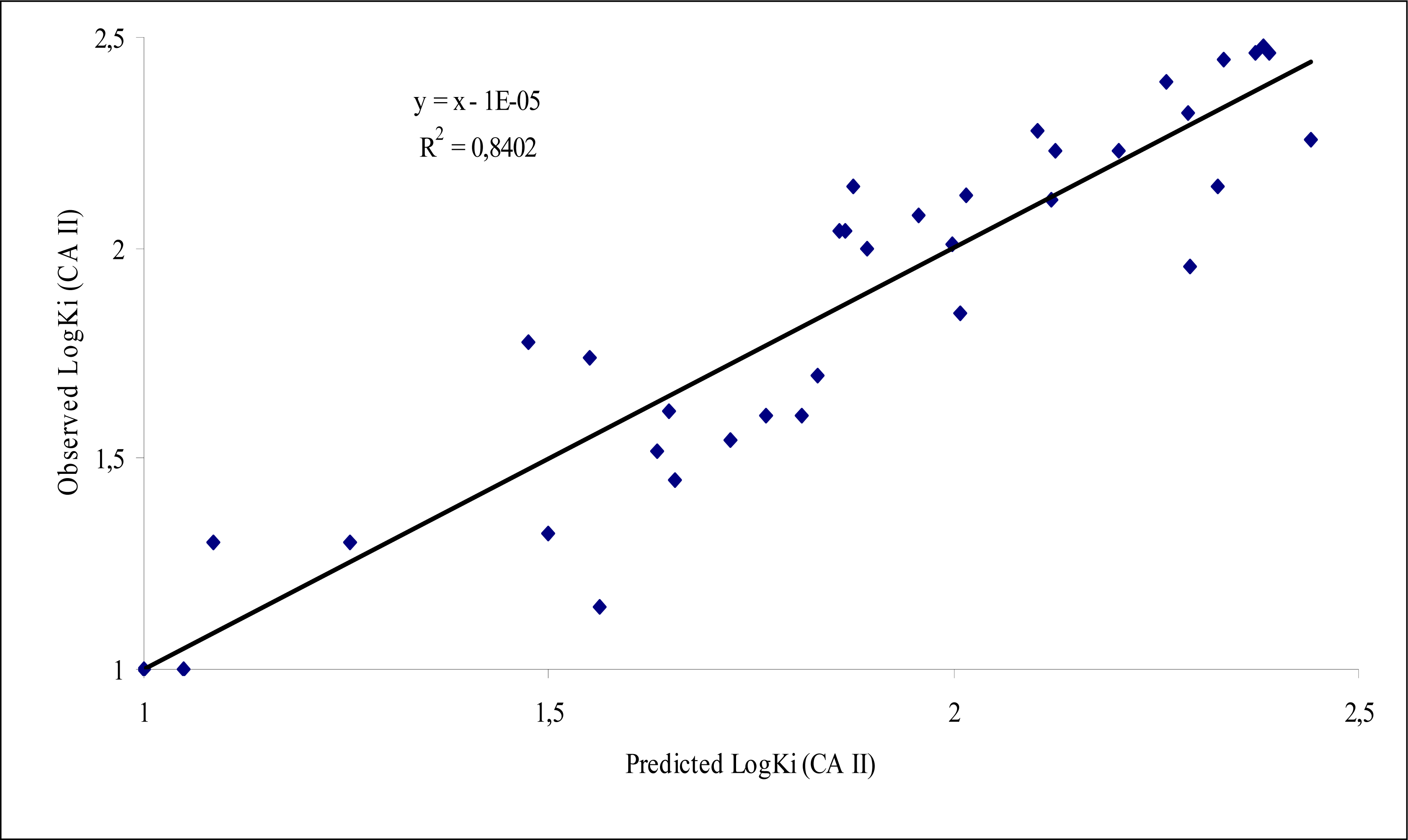
| Equ. 5 | NBR Number of benzene rings | 7.826 | 1.739(0.222) | -2.205 | 36 | 0.840 | 0.777 | 31.54 | 0.034 |
| NCA Number of C atoms | -7.510 | -0.279(0.037) | (0.940) | ||||||
| NNA Number of N atoms | 3.578 | 0.182(0.051) | |||||||
| ALP Average logP | 3.609 | 0.282(0.078) | |||||||
| 2AIC Average Information content (order 2) | 3.488 | 0.977(0.280) | |||||||
| RNBR | NBR | NCA | RNDB | DPSA-1 | MiPCN | ALS | 3χ | RPCG | NNA | ALP | 2AIC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNBR | 1.000 | |||||||||||
| NBR | 0.6504 | 1.000 | ||||||||||
| NCA | -0.1979 | 0.5717 | 1.000 | |||||||||
| RNDB | -0.3454 | -0.3573 | 0.0337 | 1.000 | ||||||||
| DPSA-1 | -0.4510 | -0.1773 | 0.2007 | -0.4079 | 1.000 | |||||||
| MiPCN | 0.4106 | 0.1693 | -0.2183 | 0.4310 | -0.4810 | 1.000 | ||||||
| ALS | -0.2483 | -0.3005 | 0.3372 | 0.6106 | 0.5991 | -0.6935 | 1.000 | |||||
| 3χ | -0.3005 | 0.4068 | -0.2031 | -0.2953 | 0.7793 | -0.0203 | -0.7565 | 1.000 | ||||
| RPCG | 0.3372 | -0.2031 | -0.7013 | 0.2939 | 0.4134 | 0.7380 | 0.2244 | -0.5260 | 1.000 | |||
| NNA | -0.6106 | -0.2953 | 0.2939 | 0.0882 | -0.1964 | 0.3227 | -0.0431 | 0.4294 | -0.3141 | 1.000 | ||
| ALP | 0.5991 | 0.7793 | 0.4134 | -0.1964 | -0.3333 | 0.2484 | -0.7649 | 0.3105 | -0.0307 | -0.3609 | 1.000 | |
| 2AIC | -0.6935 | -0.0203 | 0.7380 | 0.3227 | 0.2484 | -0.2802 | -0.3572 | 0.7993 | -0.5658 | 0.6052 | -0.1155 | 1.000 |
Share and Cite
Eroglu, E. Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors. Int. J. Mol. Sci. 2008, 9, 181-197. https://doi.org/10.3390/ijms9020181
Eroglu E. Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors. International Journal of Molecular Sciences. 2008; 9(2):181-197. https://doi.org/10.3390/ijms9020181
Chicago/Turabian StyleEroglu, Erol. 2008. "Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors" International Journal of Molecular Sciences 9, no. 2: 181-197. https://doi.org/10.3390/ijms9020181
APA StyleEroglu, E. (2008). Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors. International Journal of Molecular Sciences, 9(2), 181-197. https://doi.org/10.3390/ijms9020181