4-Chloro-2,3,5-trifluorobenzoic Acid

Abstract
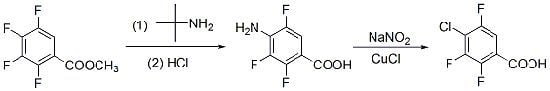
:
{kind=link}
{kind=link}
1. Introduction
2. Result and Discussion
3. Experimental
3.1. General
3.2. Synthesis of 4-Chloro-2,3,5-trifluorobenzoic Acid (1)
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sato, M.; Kawakami, H.; Motomura, T.; Aramaki, H.; Matsuda, T.; Yamashita, M.; Ito, Y.; Matsuzaki, Y.; Yamataka, K.; Ikeda, S.; et al. Quinolone carboxylic acids as a novel monoketo acid class of human immunodeficiency virus type 1 integrase inhibitors. J. Med. Chem. 2009, 52, 4869–4882. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, M.; Shah, S.S.A.; Najam, T.; Ahmad, M.M.; Tabassum, R.; Rivera, G. Synthetic Thioamide, Benzimidazole, Quinolone and Derivatives with Carboxylic Acid and Ester Moieties: A Strategy in the Design of Antituberculosis Agents. Curr. Med. Chem. 2014, 21, 911–931. [Google Scholar] [CrossRef] [PubMed]
- Lesher, G.Y. Tricyclic-Pyridinylquinoline Compounds, Their Preparation and Use. U.S. Patent 4839355 A, 13 June 1989. [Google Scholar]
- Hodgson, H.H. The sandmeyer reaction. Chem. Rev. 1947, 40, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Berrouard, P.; Dufresne, S.; Pron, A.; Veilleux, J.; Leclerc, M. Low-cost synthesis and physical characterization of thieno[3,4-c]pyrrole-4,6-dione-based polymers. J. Org. Chem. 2012, 77, 8167–8173. [Google Scholar] [CrossRef] [PubMed]

© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Feng, X.; Zhou, W.; Xia, Z.; Zhang, M.; Chen, Y.; Chen, Z. 4-Chloro-2,3,5-trifluorobenzoic Acid. Molbank 2015, 2015, M871. https://doi.org/10.3390/M871
Yu S, Feng X, Zhou W, Xia Z, Zhang M, Chen Y, Chen Z. 4-Chloro-2,3,5-trifluorobenzoic Acid. Molbank. 2015; 2015(4):M871. https://doi.org/10.3390/M871
Chicago/Turabian StyleYu, Shuitao, Xiaohu Feng, Weiyou Zhou, Zhengjun Xia, Mingguang Zhang, Yang Chen, and Zaixin Chen. 2015. "4-Chloro-2,3,5-trifluorobenzoic Acid" Molbank 2015, no. 4: M871. https://doi.org/10.3390/M871