Abstract
Direct 2(5H)-furanone annulation produces promising cross-coupling partners incorporating m- or p-bromo- and p-tosyloxyphenyl groups into the 5-position of a notable 2(5H)-furanone pharmacore. The present one-pot annulation method involves two distinctive reactions: (i) a powerful and crossed Ti-direct aldol addition and (ii) an acid-induced characteristic cyclo-condensation, leading to 2(5H)-furanones. Suzuki-Miyaura cross-coupling of 5-(4-bromophenyl)-furan-2(5H)-ones, 5-(4-tosyloxyphenyl)-3,4-dimethylfuran-2(5H)-ones and a furan derivative successfully afforded the corresponding products with the 2(5H)-furanone skeleton.
1. Introduction
2(5H)-Furanones (α,β-butenolides) are a well-recognized heterocyclic compound incorporated in natural products [1,2,3]. They also serve as useful precursors for the synthesis of lactones and furans by catalytic hydrogenation and hydride-reduction (LAH or DIBAL), respectively [2,3]. Several synthetic methods have been developed to date due to the relatively simple but promising candidate roles for 2(5H)-furanones as new pharmacores.
Current drug discovery is considerably based on cross-coupling methodologies, especially Suzuki-Miyaura cross-couplings, because large boronic acid libraries are supplied in a commercial base. Because this privileged approach representatively requires aromatics with leaving groups (-Br and -OTs) as latent scaffolds in various organic molecules, this background led us to investigate the convenient synthesis of attractive cross-coupling partner, 2(5H)-furanones 4 bearing bromophenyl and tosyloxyphenyl leaving groups.
A literature survey using SciFinder® revealed a two recent syntheses of 4a; (i) Ru-catalyzed CO insertion into the corresponding allenic alcohol [4]; and (ii) SeBr-resin-mediated lactonization of β,γ-unsaturated carboxylic acids, followed by three oxidation (H2O2)‒elimination‒methylation (MeI/LDA) reaction sequences [5]. The first method requires three steps and special handling techniques and/or apparatuses using CO. The second synthetic method requires a less accessible SeBr-resin reagent and four steps. Other target molecules 4b–4f are novel compounds.
Consistent with our longstanding studies on TiCl4/amine-mediated Claisen condensations (Ti-Claisen condensation) [6,7,8,9,10,11], its asymmetric version [12], related Ti-direct aldol reactions [13,14], and Ti-direct Mannich reactions [15], we already reported both a two-step method and a much improved one-step synthetic method for di- or trialkyl-substituted 2(5H)-furanones (direct 2(5H)-furanone or butenolide annulation) based on a TiCl4-promoted direct aldol condensation between ketones and α,α-dimethoxyketones [16,17].
2. Results and Discussion
Crossed aldol reaction between acetophenones or propiophenones and carbonyl acceptors is pivotal in organic syntheses. Ti-direct aldol additions possess considerably powerful C-C bond forming ability allowing for the reaction between less reactive different ketones which would proceed with difficulty using other reaction systems [13]. Mukaiyama, Iwasawa, and their coworkers disclosed direct crossed aldol addition between different ketones mediated by Sn(OTf)2/N-ethylpiperidine reagent, however, being limited to the reaction between both aromatic ketones [18].
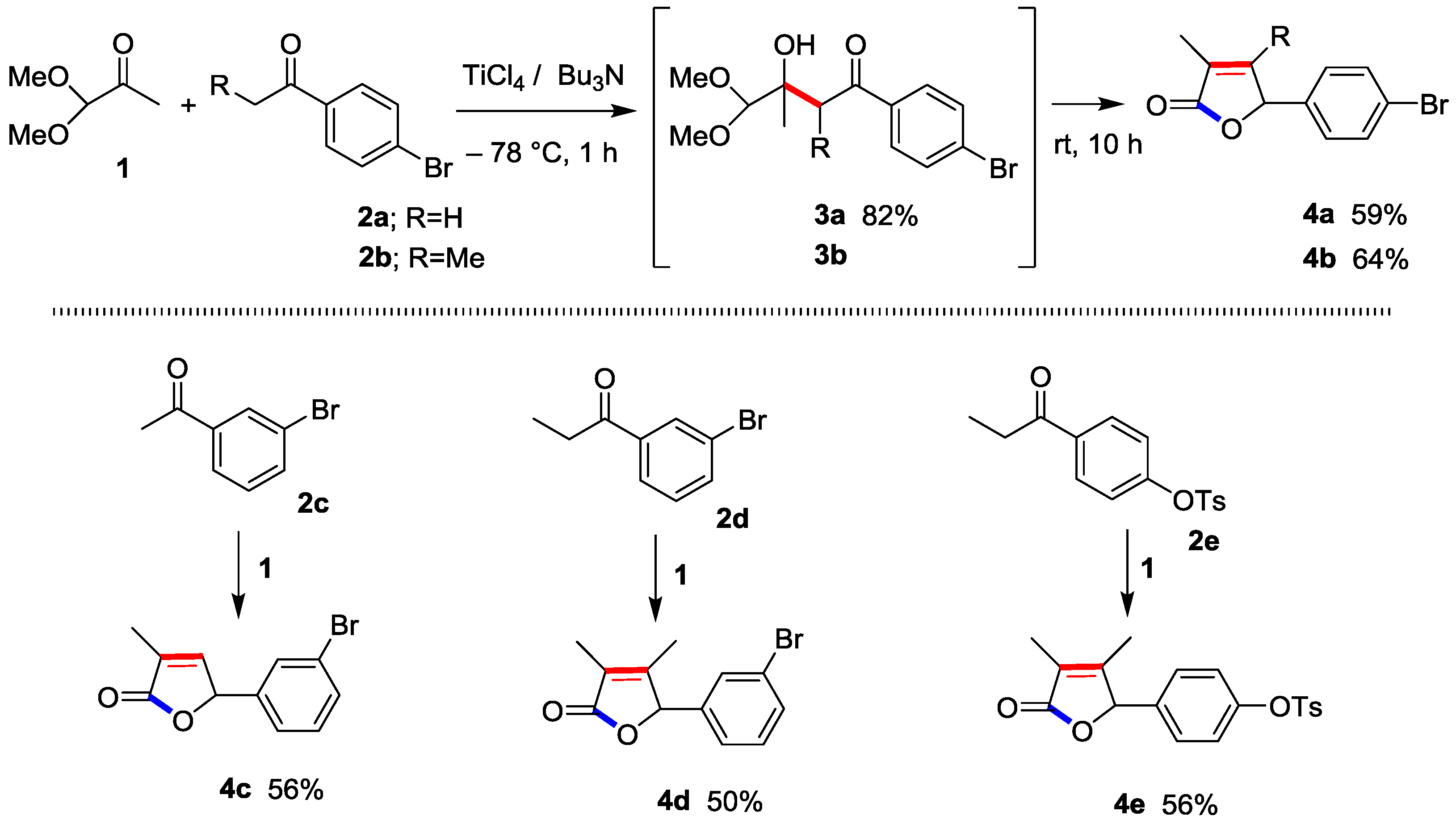
In practice, Ti-direct aldol addition 1-(4-bromophenyl)ethan-1-one (p-bromoacetophenone) (2a) with 1,1-dimethoxypropan-2-one (1,1-dimethoxyacetone) (1) proceeded smoothly to produce the aldol adduct 3a at −78 °C for 1 h in 82% isolated yield (Scheme 1). Gratifyingly, the reaction at −78 °C for 1 h and r.t. for 14 h produced 2(5H)-furanone 4a directly in 59% yield in a one-pot procedure. The direct method using 4-bromophenyl-1-ethanone (p-bromopropiophenone) (2b) instead of 2a similarly afforded the corresponding 2(5H)-furanone 4b in 64% yield through 3b under the identical conditions.
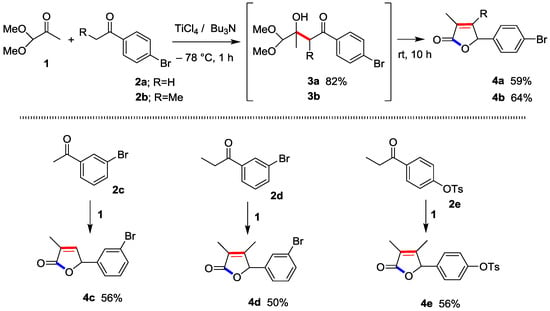
Scheme 1.
Crossed Ti-direct aldol addition and successive cyclo-condensation (furanone annulation) leading to 2(5H)-furanone 4.
The reactions using m-bromoacetophenone (2c) and m-bromopropiophenone (2d) with 1 successfully afforded the corresponding 2(5H)-furanones 4c and 4d. In addition, 2(5H)-furanone 4e bearing p-TsO- group as the related cross-coupling leaving group was also produced.
The present direct annulation methods utilize commercially available, inexpensive substrates and reagents, under accessible reaction conditions without any use of special apparatus and technique with sufficient substrate-generality [16,17]. Moreover, an application to the most straightforward total syntheses of (R)-mintlactone (a single step, 52%) and (R)-menthofuran (2 steps, overall 46%) was successfully performed [17]. Since the publication of these reports, three subsequent syntheses of (R)-mintlactone have been presented; however, these methods require (i) 3 steps, overall 7% [19]; (ii) formal synthesis, over 3 steps, accurate overall yield is unknown [20]; and (iii) 10 steps, overall 22%, [21]. The report [19] does not strictly evaluate our work [17], because it is categorized into racemic mintlactone synthesis in the authors’ reference part. The other reports [20,21] fail to refer to our work [17].
On the other hand, Mehta and Ramesh applied the present method exquisitely for the total syntheses of a series of lindenane-type sesquiterpenoids [22] and atractylenolide-type eudesmanolides [23], wherein these key skeletons are smoothly constructed, utilizing this direct 2(5H)-furanone annulation.
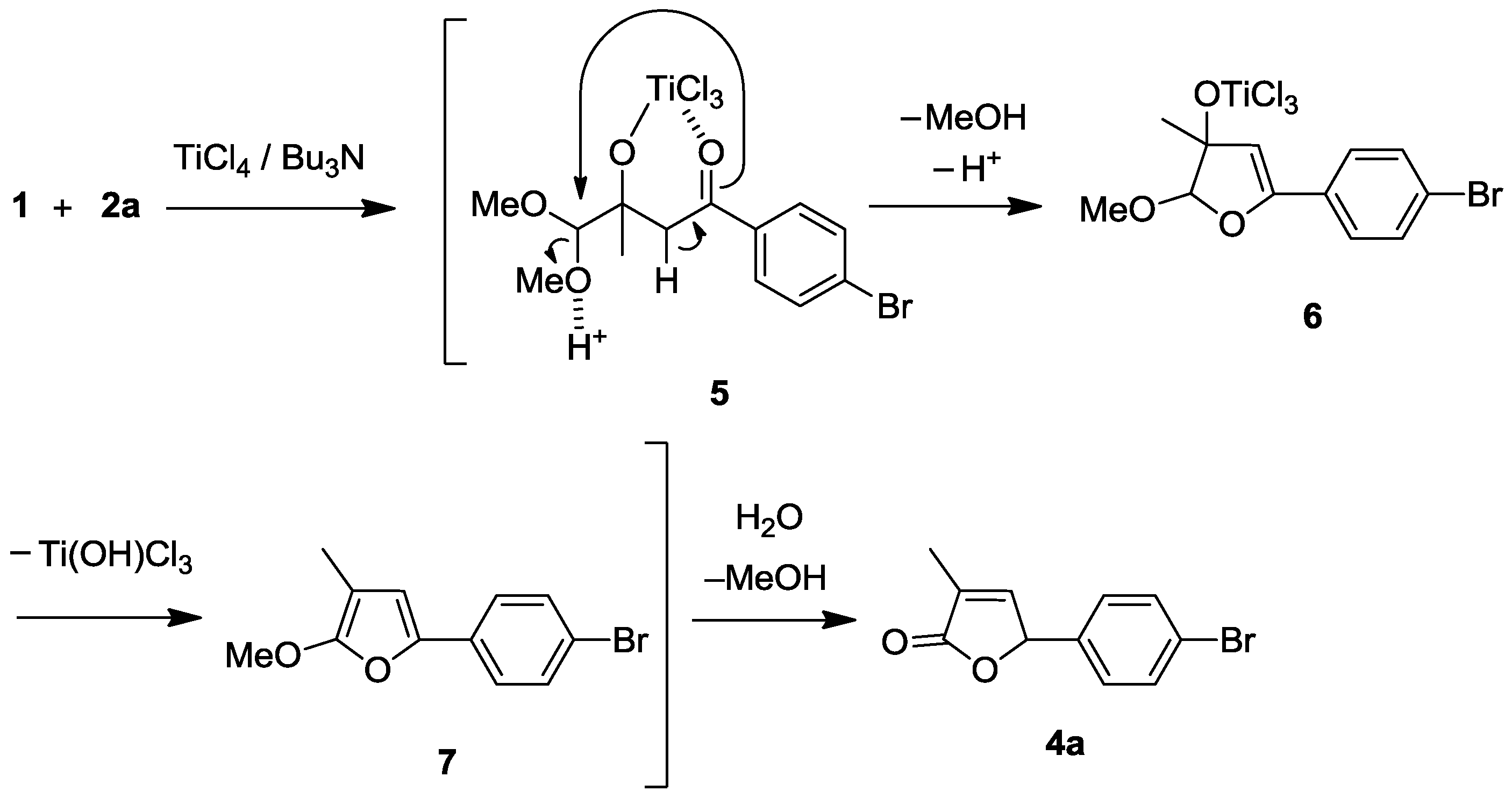
A plausible mechanism for the reaction sequences is proposed, exemplified by the production of 4a (Scheme 2). The initially formed Ti-chelated cross aldol adduct 5 is transformed to dihydrofuran intermediate 6 with ring formation and elimination of MeOH. Intermediate 6 is converted to 4a through methoxyfuran 7 with elimination of Ti(OH)Cl3 before H2O-workup.
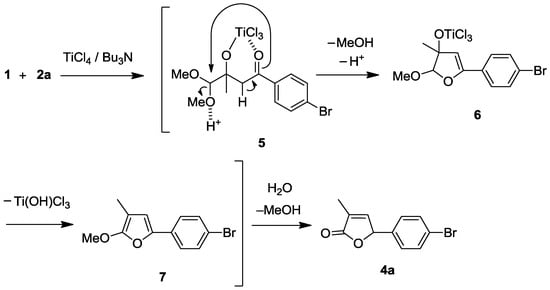
Scheme 2.
Plausible mechanism for crossed Ti-direct aldol addition and successive cyclo-condensation (furanone annulation).
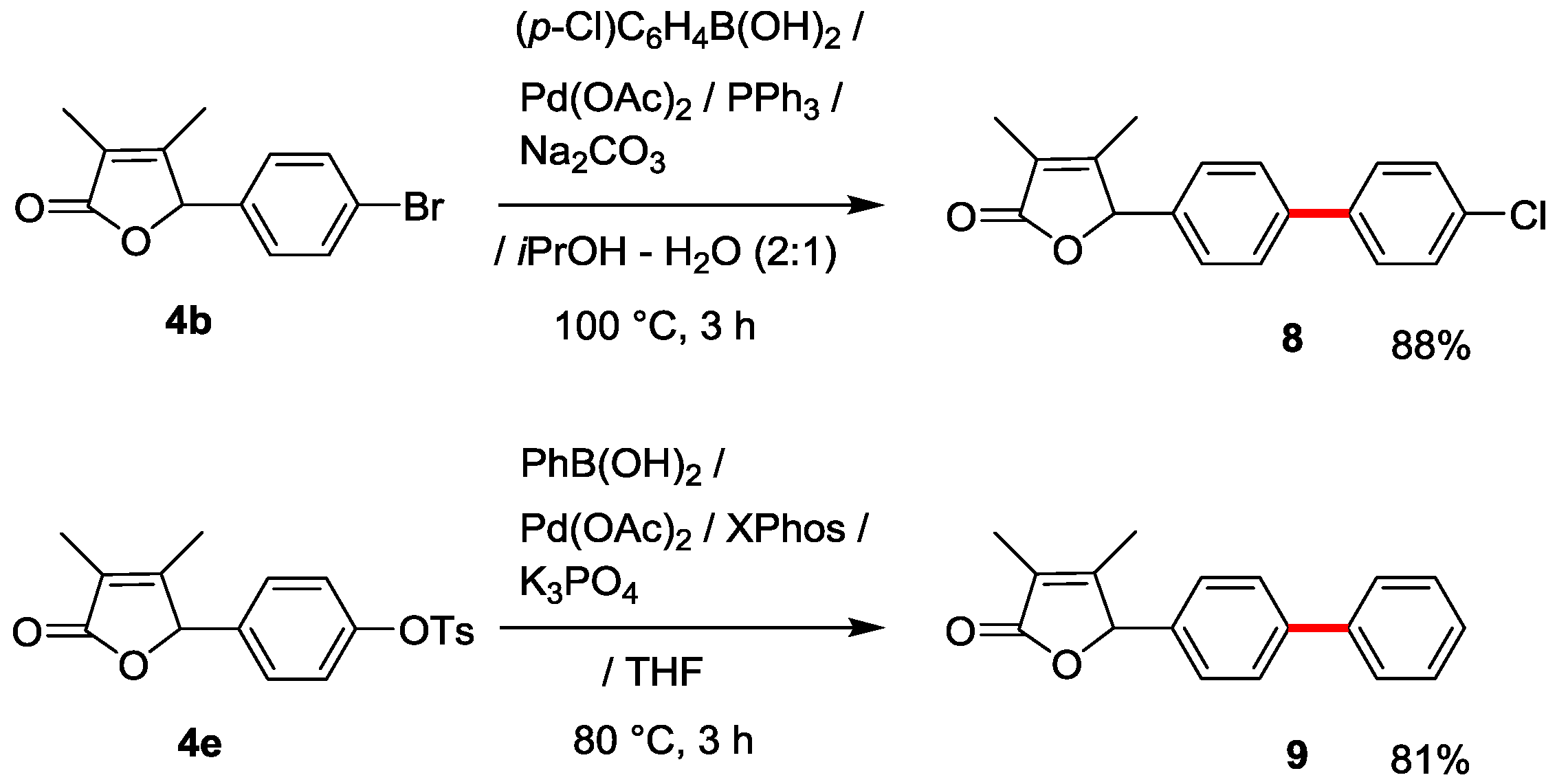
Finally, a useful derivatization of 2(5H)-furanones 4b and 4e by utilizing Suzuki-Miyaura cross-coupling was investigated (Scheme 3). A standard and accessible method using (p-Cl)C6H4B(OH)2 with Pd(OAc)2/PPh3/K2CO3 catalysis could be successfully applied to the synthesis of biphenyl-substituted 2(5H)-furanone 8 in good 88% yield. p-Tosyloxy-substituted 2(5H)-furanones 4e was also transformed to the desired compound 9 in 81% yield by utilizing Buchwald group’s modified Pd(OAc)2/XPhos/K3PO4 catalysis [24].
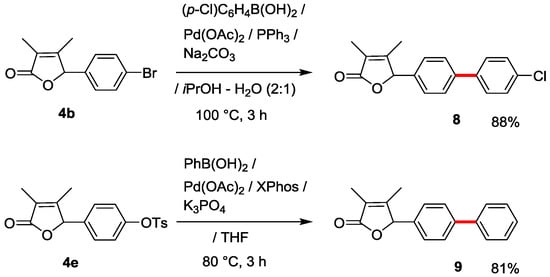
Scheme 3.
Derivatization of 2(5H)-furanones 4b and 4e by utilizing Suzuki-Miyaura cross-coupling.
Notably, furan 10 derived from 4b by DIBAL reduction (83%), similarly underwent Suzuki-Miyaura cross-coupling to produce the furan analogue 11 in 71% yield (Scheme 4).
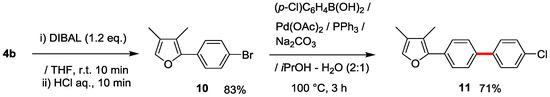
Scheme 4.
Derivatization of furan 10 derived from 4b by utilizing Suzuki-Miyaura cross-coupling.
3. Experimental Section
3.1. General
All reactions were carried out in oven-dried glassware under an argon atmosphere. Flash column chromatography was performed with Silica Gel 60 (spherical) (63–210 μm, KANTO CHEMICAL, Tokyo, Japan). TLC analysis was performed on 0.25 mm Silicagel Merck 60 F254 plates. Melting points were determined on a hot stage microscope apparatus (ATM-01, AS ONE, Osaka, Japan) and were uncorrected. NMR spectra were recorded on a JEOLRESONANCE ECX-500II spectrometer (JEOL, Tokyo, Japan) operating at 500 MHz for 1H-NMR and 125 MHz for 13C-NMR. Chemical shifts (δ ppm) in CDCl3 were reported downfield from TMS (= 0.00) for 1H-NMR. For 13C-NMR, chemical shifts were reported in the scale relative to CDCl3 (77.0 ppm) as an internal reference. IR Spectra were recorded on a SHIMADZU IRAffinity-1S spectrophotometer (SHIMADZU, Kyoto, Japan). Mass spectra were measured on a JEOL JMS-T100LCP spectrometer (JEOL, Tokyo, Japan).
3.2. Preparation of 1-(4-Bromophenyl)-3-hydroxy-4,4-dimethoxy-3-methylbutan-1-one (3a)
TiCl4 (0.82 mL, 7.5 mmol) and Bu3N (2.38 mL, 10.0 mmol) were successively added to a stirred solution of p-bromoacetophenone (2a; 1.00 g, 5.0 mmol) in CH2Cl2 (5.0 mL) at −78 °C under an Ar atmosphere. After 15 min, 1,1-dimethoxyacetone (1; 1.18 g, 10.0 mml) in CH2Cl2 (5.0 mL) was added to the mixture at −78 °C, followed by being stirred at the same temperature for 1 h. Water was added to the mixture, which was extracted twice with Et2O. The combined organic phase was washed with 1M HCl aq., brine, dried (Na2SO4), and concentrated. The obtained crude product (1.97 g) was purified by SiO2-column chromatography (hexane/AcOEt = 4:1) to give the desired product 3a (1.31 g, 82 %).
Pale yellow oil; 1H-NMR (500 MHz, CDCl3): δ = 1.30 (s, 3H), 2.87 (d, J = 16.0 Hz, 1H), 3.36 (d, J = 16.0 Hz, 1H), 3.47 (s, 3H), 3.51 (s, 3H), 4.03 (s, 1H), 4.16 (s, 1H), 7.59–7.62 (m, 2H), 7.82–7.85 (m, 2H); 13C-NMR (125 MHz, CDCl3): δ = 23.4, 42.3, 57.9, 75.3, 110.1 (2C), 128.3, 129.8 (2C), 131.7 (2C), 136.3, 200.7; IR (neat): νmax = 3482, 2936, 2832, 1736, 1672, 1583, 1396, 1219, 1184 cm−1; HRMS (ESI): m/z calcd for C13H17O4Br [M + Na]+ 339.0208; found: 339.0205.
3.3. Preparation of 5-(4-Bromophenyl)-3-methylfuran-2(5H)-one (4a) [4]
TiCl4 (0.82 mL, 7.5 mmol) and Bu3N (2.38 mL, 10.0 mmol) were successively added dropwise to a stirred solution of p-bromoacetophenone (2a; 1.00 g, 5.0 mmol) in CH2Cl2 (5.0 mL) at −78 °C under an Ar atmosphere. After 15 min, 1,1-dimethoxyacetone (1; 1.18 g, 10.0 mml) in CH2Cl2 (5.0 mL) was added. Then the reaction mixture was stirred at the same temperature for 1 h and 20–25 °C for 14 h. Water was added to the mixture, which was extracted twice with Et2O. The combined organic phase was washed with 1M HCl, brine, dried (Na2SO4), and concentrated. The obtained crude product (2.22 g) was purified by SiO2-column chromatography (hexane/AcOEt = 5:1) to give the desired product 4a (0.74 g, 59 %).
Pale yellow crystals (recryst. from iPrOH); mp 93–94 °C (lit. 94.0 °C [4]); 1H-NMR (500 MHz, CDCl3): δ = 2.00 (t, J = 1.7 Hz, 3H), 5.82 (quin, J = 1.7 Hz, 1H), 7.09 (quin, J = 1.7 Hz, 1H), 7.14 (d, J = 8.6 Hz, 2H), 7.52 (d, J = 8.6 Hz, 2H); 13C-NMR (125 MHz, CDCl3): δ = 10.6, 81.3, 123.1, 128.0 (2C), 129.8, 132.0 (2C), 134.2, 147.8, 173.9; IR (neat): νmax = 3084, 2928, 2363, 1748, 1661, 1587, 1485, 1406, 1319, 1294 cm−1.
3.4. Preparation of 5-(4-Bromophenyl)-3,4-dimethylfuran-2(5H)-one (4b)
According to a similar procedure for preparing 4a, the reaction of p-bromopropiophenone (2b; 1.07 g 5.0 mmol) with 1,1-dimethoxyacetone (1; 1.18 g, 10.0 mml), using TiCl4 (0.82 mL, 7.5 mmol) and Bu3N (2.38 mL, 10.0 mmol) in CH2Cl2 (10.0 mL) gave the desired product (4b; 0.86 g, 64 %).
Pale yellow crystals (recryst. from iPrOH); mp 112–114 °C; 1H-NMR (500 MHz, CDCl3): δ = 1.81 (s, 3H), 1.89 (s, 3H), 5.56 (s, 1H), 7.09 (d, J = 8.6 Hz, 2H), 7.52 (d, J = 8.6 Hz, 2H); 13C-NMR (125 MHz, CDCl3): δ = 8.6, 12.1, 84.2, 123.2, 123.3, 128.4 (2C), 132.1 (2C), 134.0, 158.6, 174.4; IR (neat): νmax = 2363, 1744, 1672, 1489, 1433, 1406, 1321, 1287 cm−1; HRMS (ESI): m/z calcd for C12H11O2Br [M + Na]+ 288.9840; found: 288.9839.
3.5. Preparation of 5-(3-Bromophenyl)-3-methylfuran-2(5H)-one (4c)
According to a similar procedure for preparing 4a, the reaction of m-bromoacetophenone (2c; 1.00 g, 5.0 mmol) with 1,1-dimethoxypropanone (1; 1.18 g, 10.0 mml), using TiCl4 (0.82 mL, 7.5 mmol) and Bu3N (2.38 mL, 10.0 mmol) in CH2Cl2 (10.0 mL) gave the desired product (4c; 0.74 g, 56 %).
Pale yellow crystals (recryst. from iPrOH); mp 97–98 °C; 1H-NMR (500 MHz, CDCl3): δ = 2.00 (t, J = 1.7 Hz, 3H), 5.83 (quin, J = 1.7 Hz, 1H), 7.11 (quin, J = 1.7 Hz, 1H), 7.21 (d, J = 7.5 Hz, 1H), 7.26 (dd, J = 7.5, 8.0 Hz, 1H), 7.39–7.42 (m, 1H), 7.49 (d, J = 7.5 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 10.6, 81.0, 122.9, 125.0, 129.3, 129.9, 130.5, 132.1, 137.4, 147.7, 173.8; IR (neat): νmax = 3063, 2357, 1748, 1661, 1593, 1566, 1471, 1429 cm−1; HRMS (ESI): m/z calcd for C11H9O2Br [M + Na]+ 274.9684; found: 274.9681.
3.6. Preparation of 5-(3-Bromophenyl)-3,4-dimethylfuran-2(5H)-one (4d)
According to a similar procedure for preparing 4a, the reaction of p-bromopropiophenone (2d; 1.07 g 5.0 mmol) with 1,1-dimethoxyacetone (1; 1.18 g, 10.0 mml), using TiCl4 (0.82 mL, 7.5 mmol) and Bu3N (2.38 mL, 10.0 mmol) in CH2Cl2 (10.0 mL) gave the desired product (4d; 0.66 g, 50 %).
Pale yellow crystals (recryst. from iPrOH); mp 77–78 °C; 1H-NMR (500 MHz, CDCl3): δ = 1.82 (s, 3H), 1.90 (s, 3H), 5.55 (s, 1H), 7.15 (d, J = 8.3 Hz, 1H), 7.27 (t, J = 8.3 Hz, 1H), 7.34–7.35 (m, 1H), 7.49–7.53 (m, 1H); 13C-NMR (125 MHz, CDCl3): δ = 8.6, 12.1, 84.0, 122.9, 123.4, 125.5, 129.6, 130.4, 132.3, 137.3, 158.6, 174.3; IR (neat): νmax = 3057, 2922, 1748, 1072, 997 cm−1; HRMS (ESI): m/z calcd for C12H11O2Br [M + Na]+ 267.0021, found 267.0023.
3.7. Preparation of 5-(4-Tosyloxyphenyl)-3,4-dimethylfuran-2(5H)-one (4e)
According to a similar procedure for preparing 4b using m-tosyloxypropiophenone (2f; 1.52 g, 5.00 mmol) with 1,1-dimethoxyacetone (1; 1.18 g, 10.0 mml), using TiCl4 (0.82 mL, 7.5 mmol) and Bu3N (2.38 mL, 10.0 mmol) in CH2Cl2 (10.0 mL) gave the desired product (4e; 1.06 g, 56%).
Colorless crystals (recryst. from iPrOH); mp 128–129 °C; 1H-NMR (500 MHz, CDCl3): δ = 1.79 (s, 3H), 1.88 (s, 3H), 2.45 (s, 3H), 5.56 (s, 1H), 6.99–7.04 (m, 2H), 7.12–7.16 (m, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H); 13C-NMR (125 MHz, CDCl3): δ = 8.5, 12.1, 21.6, 84.0, 122.9 (2C), 123.3, 128.1 (2C), 128.4 (2C), 129.8 (2C), 132.0, 133.9, 145.6, 150.0, 158.6, 174.3; IR (neat): νmax = 1736, 1501, 1371, 1177, 1153, 1090, 1007, 868, 716 cm−1; HRMS (ESI): m/z calcd for C19H18O5S [M + Na]+ 381.0773; found: 381.0783.
3.8. Synthesis of 5-(4’-Chloro-[1,1’-biphenyl]-4-yl)-3,4-dimethylfuran-2(5H)-one (8)
A mixture of 4b (134 mg, 0.50 mmol), (p-Cl)C6H4B(OH)2 (82 mg, 0.53 mmol), Pd(OAc)2 (1.1 mg, 0.005 mmol), PPh3 (3.9 mg, 0.015 mmol), and Na2CO3 (64 mg, 0.60 mmol) in iPrOH/H2O (2:1, 1.4 mL) was stirred at 100–105 °C for 3 h under an Ar atmosphere. After cooling down to r.t., water was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4), and concentrated. The obtained crude product (165 mg) was purified by SiO2-column chromatography (hexane/AcOEt = 5:1) to give the desired product 8 (131 mg, 88%).
Colorless crystals (recryst. from iPrOH); mp 157–158 °C; 1H-NMR (500 MHz, CDCl3): δ = 1.85 (s, 3H), 1.91 (s, 3H), 5.64 (s, 1H), 7.25–7.30 (m, 2H), 7.39–7.43 (m, 2H), 7.48–7.52 (m, 2H), 7.54–7.58 (m, 2H); 13C-NMR (125 MHz, CDCl3): δ = 8.6, 12.1, 84.7, 123.1, 127.3 (2C), 127.4 (2C), 128.2 (2C), 128.9 (2C), 133.7, 134.2, 138.6, 140.8, 159.0, 174.6; IR (neat): νmax = 1748, 1674, 1483, 1319, 1080, 1013, 814, 760, 665 cm−1; HRMS (ESI): m/z calcd for C18H15O2Cl [M + H]+ 299.0839; found: 299.0864.
3.9. Synthesis of 5-(1,1’-Biphenyl-4-yl)-3,4-dimethylfuran-2(5H)-one (9) [25]
A mixture of 4f (179 mg, 0.50 mmol), PhB(OH)2 (122 mg, 1.00 mmol), Pd(OAc)2 (2.2 mg, 0.01 mmol), XPhos (11.9 mg, 0.025 mmol), and K3PO4 (318 mg, 1.50 mmol) in THF (1.0 mL) was stirred at 80–85 °C for 3 h under an Ar atmosphere. A similar work up for the synthesis of 8 gave the desired product 9 (107 mg, 81%).
Yellow solid (recryst. from iPrOH); mp 121–122 °C; 1H-NMR (500 MHz, CDCl3): δ = 1.86 (s, 3H), 1.92 (s, 3H), 5.65 (s, 1H), 7.28 (d, J = 8.6 Hz, 2H), 7.34–7.39 (m, 1H), 7.42–7.48 (m, 2H), 7.56–7.63 (m, 4H); 13C-NMR (125 MHz, CDCl3): δ = 8.6, 12.2, 84.8, 123.2, 127.1 (2C), 127.2 (2C), 127.6 (2C), 128.8 (2C), 133.9, 140.3, 142.2, 159.0, 174.7; IR (neat): νmax = 1742, 1678, 1086, 993, 756, 694 cm−1; HRMS (ESI): m/z calcd for C18H16O2 [M + Na]+ 287.1048; found: 287.1038.
3.10. Synthesis of 2-(4-Bromopheyl)-3,4-dimethylfuran (10)
DIBAL (1.0 M in toluene; 0.60 mL, 0.60 mmol) was added dropwise to a stirred solution of 4b (134 mg, 0.50 mmol) in THF (1.5 mL) at 0–5 °C under an Ar atmosphere. After 10 min, 1M-HCl aq. solution was added to the mixture at 0–5 °C, followed by being stirred at the same temperature for 10 min. Water was added to the mixture, which was extracted with Et2O (×2). The combined organic phase was washed with 1M-HCl aq. solution (×2), NaHCO3 aq. solution (×2), brine, dried (Na2SO4) and concentrated. The obtained crude product (164 mg) was purified by SiO2 (Note: “60 N spherical, neutral” was used instead of standard type) -column chromatography (hexane/AcOEt = 5:1) to give the desired product 10 (105 mg, 83%).
Colorless solid; mp 44–46 °C; 1H-NMR (500 MHz, CDCl3): δ = 1.99 (s, 3H), 2.16 (s, 3H), 7.21 (s, 1H), 7.45–7.54 (m, 4H); 13C-NMR (125 MHz, CDCl3): δ = 8.3, 9.6, 117.5, 120.3, 122.9, 126.7 (2C), 131.0, 131.5 (2C), 138.0, 147.7; IR (neat): νmax = 2922, 2866, 1545, 1483, 1393, 1074, 1003, 896, 824 cm−1; HRMS (DART): m/z calcd for C12H11OBr [M + H]+ 251.0072; found: 251.0068.
3.11. Synthesis of 2-(4’-Chloro-[1,1’-biphenyl]-4-yl)-3,4-dimethylfuran (11)
The mixture of 10 (126 mg, 0.50 mmol), (p-Cl)C6H4B(OH)2 (82 mg, 0.53 mmol), Pd(OAc)2 (1.1 mg, 0.005 mmol), PPh3 (3.9 mg, 0.015 mmol), and Na2CO3 (64 mg, 0.60 mmol) in iPrOH/H2O (2:1, 1.4 mL) was stirred at 100–105 °C for 3 h under an Ar atmosphere. After cooling down to r.t., water was added to the mixture, which was extracted with AcOEt (×2). The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product (166 mg) was purified by SiO2 (Note: “60 N spherical, neutral” was used instead of standard type)-column chromatography (hexane/AcOEt = 5:1) to give the desired product 11 (100 mg, 71%).
Pale green solid (recryst. from iPrOH); mp 126–129 °C; 1H-NMR (500 MHz, CDCl3): δ = 2.01 (s, 3H), 2.22 (s, 3H), 7.24 (s, 1H7.41 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 8.6 Hz, 2H), 7.59 (d, J = 8.6 Hz, 2H), 7.69 (d, J = 8.6 Hz, 2H); 13C-NMR (125 MHz, CDCl3): δ = 8.4, 9.7, 117.4, 122.9, 125.6 (C), 126.9 (2C), 128.1 (2C), 128.9 (2C), 131.4, 133.3, 137.8, 137.9, 139.1, 148.3; IR (neat): νmax = 2920, 2860, 1481, 1096, 1001, 816, 718 cm−1; HRMS (DART): m/z calcd for C18H15OCl [M + H]+ 283.0890; found: 283.0890.
4. Conclusions
We developed an expedient synthetic approach to 5-(m- or p-bromo- and p-tosyloxyphenyl)-3-methyl-furan-2(5H)-ones and its 3,4-dimethyl analogues utilizing TiCl4-mediated 2(5H)-furanone annulation. These molecules comprise a fundamentally important 2(5H)-furanone skeleton with m- or p-leaving moiety as a basic probe for the cross-coupling partner. Suzuki-Miyaura cross-coupling was successfully applied for p-bromo and p-tosyloxy substrates to afford the corresponding biphenyl products. In addition, a furan derivative prepared from a 2(5H)-furanone by DIBAL reduction underwent smoothly Suzuki-Miyaura cross-coupling to give the biphenyl analogue. The utility of the Ti-Claisen and the crossed Ti-direct aldol reactions is also demonstrated in recent asymmetric total syntheses of alternaric acid [11] and azaspirene [26].
Supplementary Materials
All materials (substrates and reagents) in this work are commercially available with inexpensive price. Copies of the 1H, 13C-NMR spectra for compounds 3a, 4a–4e, 8–11 are available in the supplementary information. They and molfiles can be found at http://www.mdpi.com/1422-8599/2016/4/M908.
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Acknowledgments
This research was partially supported by Grant-in-Aids for Scientific Research on Basic Areas (B) “18350056” and (C) 15K05508, Priority Areas (A) “17035087” and “18037068”, and Exploratory Research “17655045” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Author Contributions
Y.B. and Y.A. contributed the whole syntheses. Y.T. prepared the whole manuscript. H.N. prepared the reference part.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marshal, P.G. Rodd’s Chemistry of Carbon Compounds, 2nd ed.; Elsevier: New York, NY, USA, 1970; Volume 2, Part D; pp. 369–375. [Google Scholar]
- Rao, Y.S. Recent advances in the chemistry of unsaturated lactones. Chem. Rev. 1976, 76, 625–694. [Google Scholar] [CrossRef]
- Knight, D.W. Synthetic approaches to butenolides. Contemp. Org. Synth. 1994, 1, 287–315. [Google Scholar] [CrossRef]
- Yoneda, E.; Zhang, S.W.; Zhou, D.Y.; Onitsuka, K.; Takahashi, S. Ruthenium-catalyzed cyclocarbonylation of allenyl alcohols and amines: Selective synthesis of lactones and lactams. J. Org. Chem. 2003, 68, 8571–8576. [Google Scholar] [CrossRef] [PubMed]
- He, R.J.; Zhu, B.C.; Wang, Y.G. Lewis base-catalyzed electrophilic lactonization of selenyl bromide resin and facile solid-phase synthesis of furan-2(5H)-one derivatives. Appl. Organometal. Chem. 2014, 28, 523–528. [Google Scholar]
- Tanabe, Y. The selective Claisen and Dieckmann ester condensations promoted by dichlorobis(trifluoromethanesulfonato)titanium (IV). Bull. Chem. Soc. Jpn. 1989, 62, 1917–1924. [Google Scholar] [CrossRef]
- Tanabe, Y.; Hamasaki, R.; Funakoshi, S. Powerful Claisen condensation and Claisen-aldol tandem reaction of α,α-disubstituted esters promoted by ZrCl4‒iPr2NEt. Chem. Commun. 2001, 1674–1675. [Google Scholar] [CrossRef]
- Tanabe, Y.; Makita, A.; Funakoshi, S.; Hamasaki, R.; Kawakusu, T. Practical synthesis of Z-Civetone utilizing Ti-Dieckmann condensation. Adv. Synth. Catal. 2002, 344, 507–510. [Google Scholar] [CrossRef]
- Misaki, T.; Nagase, R.; Matsumoto, K.; Tanabe, Y. Ti-crossed Claisen condensation between carboxylic esters and acid chlorides or acids: Highly selective and general method for the preparation of various β-keto esters. J. Am. Chem. Soc. 2005, 127, 2854–2855. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Nakazawa, S.; Okabayashi, T.; Horii, A.; Misaki, T.; Tanabe, Y. Powerful Ti-crossed Claisen condensation between ketene silyl acetals or thioacetals and acid chlorides or acids. Org. Lett. 2006, 8, 5215–5218. [Google Scholar] [CrossRef] [PubMed]
- Nagase, R.; Oguni, Y.; Ureshino, S.; Mura, H.; Misaki, T.; Tanabe, Y. Asymmetric Ti-crossed Claisen condensation: Application to concise asymmetric total synthesis of Alternaric acid. Chem. Commun. 2013, 7001–7003. [Google Scholar] [CrossRef] [PubMed]
- Ashida, Y.; Kajimoto, S.; Nakatsuji, H.; Tanabe, Y. Synthesis of methyl 1-formylcyclopropanecarboxylate utilizing Ti-Claisen condensation. Org. Synth. Accepted paper.
- Tanabe, Y.; Matsumoto, N.; Higashi, T.; Misaki, T.; Itoh, T.; Yamamoto, M.; Mitarai, K.; Nishii, Y. Direct, practical, and powerful crossed-aldol additions between ketones and ketones or aldehydes utilizing environmentally benign TiCl4–Bu3N reagent. Tetrahedron (Symposium) 2002, 58, 8269–8280. [Google Scholar] [CrossRef]
- Nagase, R.; Matsumoto, N.; Hosomi, K.; Higashi, T.; Funakoshi, S.; Misaki, T.; Tanabe, Y. Ti-direct, powerful, stereoselective aldol-type additions of esters and thioesters to carbonyl compounds: Application to the synthesis and evaluation of lactone analogs of Jasmone perfumes. Org. Biomol. Chem. 2007, 5, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Funatomi, T.; Nakazawa, S.; Matsumoto, K.; Nagase, R.; Tanabe, Y. Ti-mediated direct and highly stereoselective Mannich reactions between esters and oxime ethers. Chem. Commun. 2008, 771–773. [Google Scholar] [CrossRef]
- Tanabe, Y.; Ohno, N. A novel and efficient synthesis of 2(5H)-furanone derivative. J. Org. Chem. 1988, 53, 1560–1563. [Google Scholar] [CrossRef]
- Tanabe, Y.; Mitarai, K.; Higashi, T.; Misaki, T.; Nishii, Y. Efficient one-step synthesis of trialkylsubstituted 2(5H)-furanone utilizing direct Ti-crossed aldol condensation and its application to the straightforward synthesis of (R)-mintlactone and (R)-menthofuran. Chem. Commun. 2002, 2542–2543. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Iwasawa, N.; Stevens, R.W.; Haga, T. Synthetic control leading to chiral compounds. Tetrahedron 1984, 40, 1381. [Google Scholar] [CrossRef]
- Gao, P.; Xu, P.-F.; Zhai, H. Expeditious construction of (+)-mintlactone via intramolecular hetero-Pauson−Khand reaction. J. Org. Chem. 2009, 74, 2592–2593. [Google Scholar] [CrossRef] [PubMed]
- Curini, M.; Epifano, F.; Montanari, F. Oxone®-KI induced lactonization and etherification of unsaturated acids and alcohols: A formal synthesis of mintlactone. Synlett 2004, 368–370. [Google Scholar] [CrossRef]
- Bates, R.W.; Scidhar, S. A synthesis of (−)-Mintlactone. J. Org. Chem. 2008, 73, 8104–8105. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Mehta, G. A total synthesis of sarcandralactone A: A general, concise, RCM enabled approach to lindenanolide sesquiterpenoids. Tetrahedron Lett. 2015, 56, 3941–3944. [Google Scholar] [CrossRef]
- Ramesh, S.; Mehta, G. A general, concise, ‘collective’ approach to eudesmanolide sesquiterpenoids: total synthesis of bioactive atractylenolides I-IV and related natural products. Tetrahedron Lett. 2015, 56, 5545–5548. [Google Scholar] [CrossRef]
- Billingsley, K.; Buchwald, S.L. Highly efficient monophosphine-based catalyst for the palladium-catalyzed suzuki-miyaura reaction of heteroaryl halides and heteroaryl boronic acids and esters. J. Am. Chem. Soc. 2007, 129, 3358–3366. [Google Scholar] [CrossRef] [PubMed]
- Lutz, R.E.; Couper, M. The reduction of cis and trans 2,3-dimethyl-3-p-xenoylacrylic acids and their esters. J. Org. Chem. 1941, 6, 91–104. [Google Scholar] [CrossRef]
- Sugi, M.; Nagase, R.; Misaki, T.; Nakatsuji, H.; Tanabe, Y. Asymmetric Total Synthesis of (–)-Azaspirene by utilizing Ti-Claisen Condensation and Ti-Direct Aldol Reaction. Eur. J. Org. Chem. in press. [CrossRef]
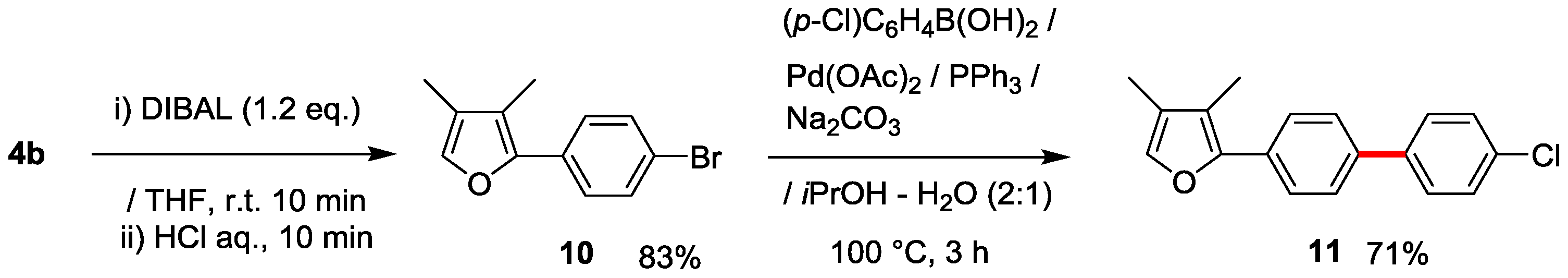
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).