1,3-Bis(2,6-diisopropylphenyl)-2-trichloromethylimidazolidine


Abstract
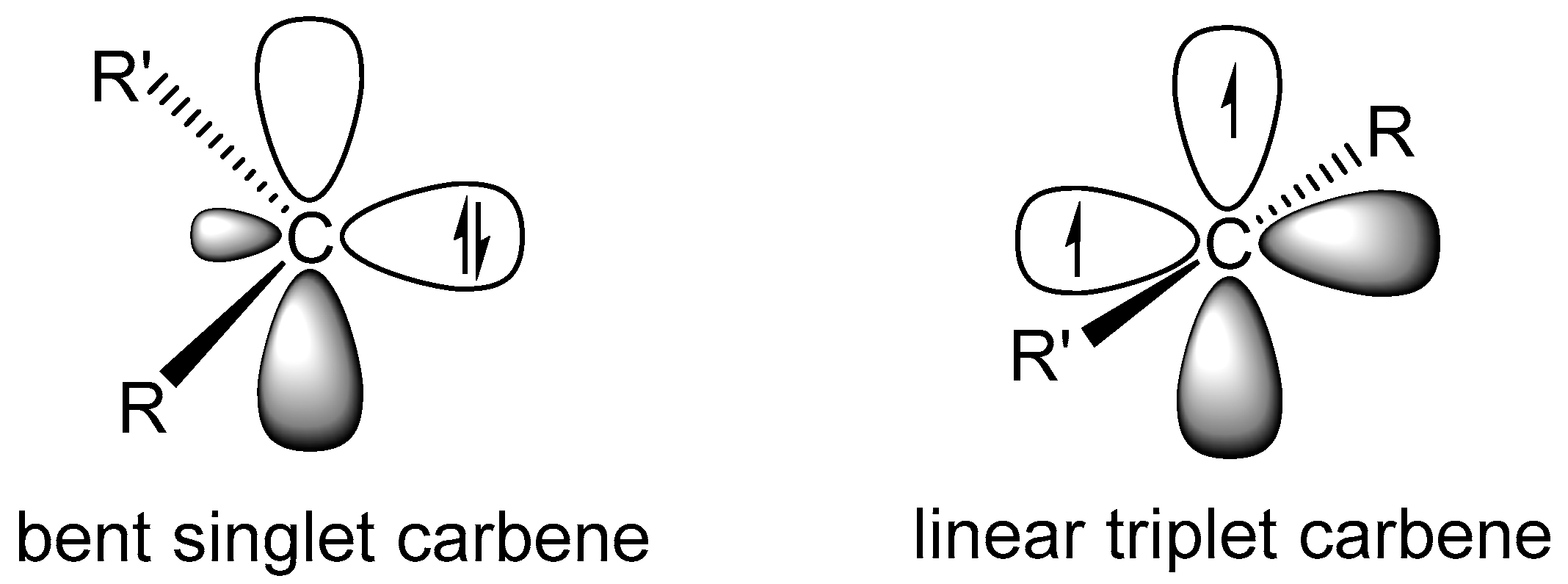
:1. Introduction
2. Results and Discussion
2.1. Synthetic Discussion
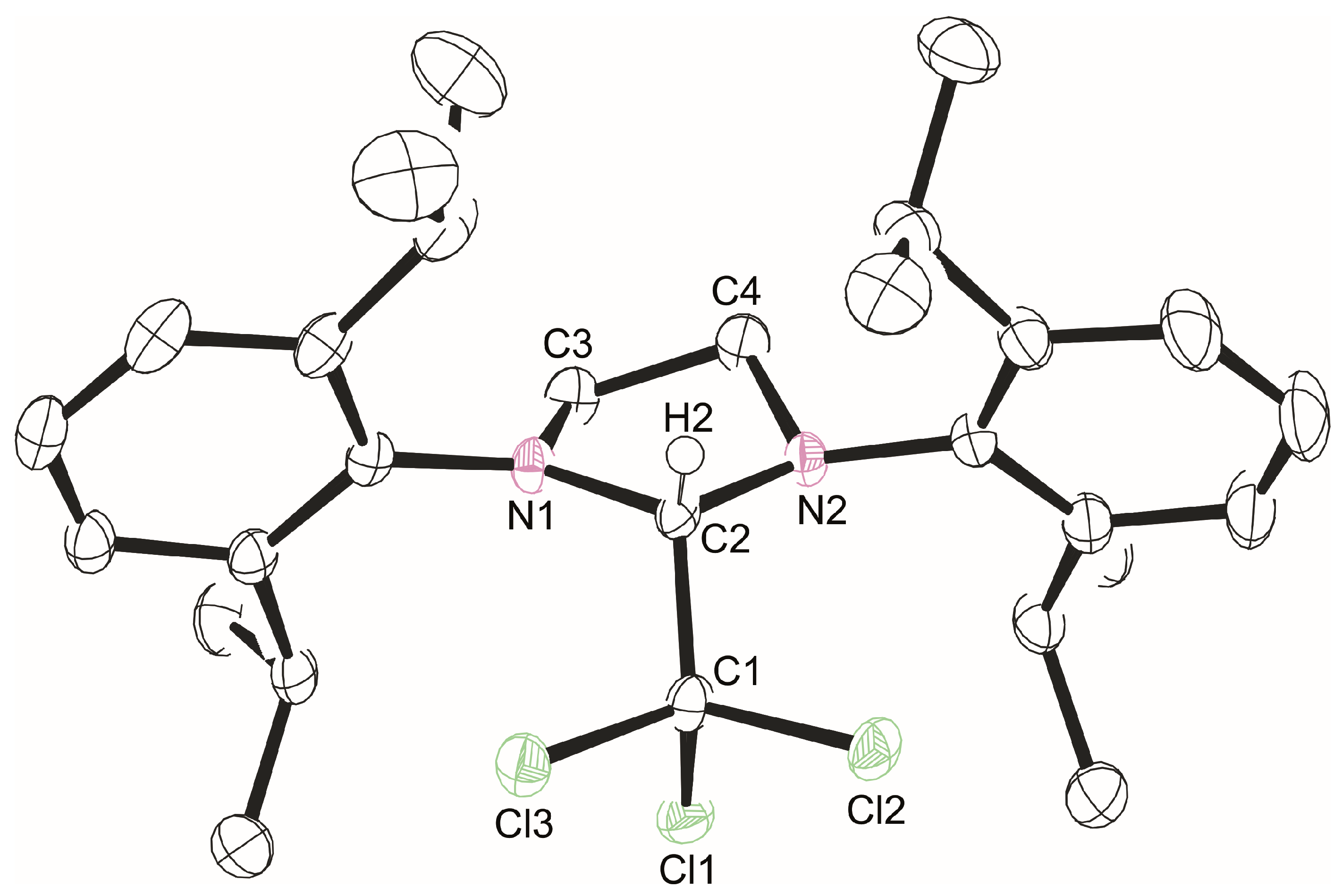
2.2. X-ray Crystallography
3. Materials and Methods
3.1. General Methods, X-ray Crystallography Sample Collection, and Computational Details
3.2. Synthesis of SIPr(H)CCl3
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bertrand, G.; Reed, R. λ3-Phosphinocarbenes λ5-phosphaacetylenes. Coord. Chem. Rev. 1994, 137, 323–355. [Google Scholar] [CrossRef]
- Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G. Analogous α,α′-bis-carbenoid triply bonded species: Synthesis of a stable λ3-phosphinocarbene-λ5-phosphaacetylene. J. Am. Chem. Soc. 1988, 110, 6463–6466. [Google Scholar] [CrossRef]
- Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Stable cyclic (alkyl)(amino)carbenes as rigid or flexible, bulky, electron-rich ligands for transition-metal catalysts: A quaternary carbon atom makes the difference. Angew. Chem. Int. Ed. 2005, 117, 5851–5855. [Google Scholar] [CrossRef]
- Frey, G.D.; Lavallo, V.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science 2007, 316, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Aldeco-Perez, E.; Rosenthal, A.J.; Donnadieu, B.; Parameswaran, P.; Frenking, G.; Bertrand, G. Isolation of a C5-deprotonated imidazolium, a crystalline “abnormal” N-heterocyclic carbene. Science 2009, 326, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Bourissou, D.; Guerret, O.; Gabbai, F.P.; Bertrand, G. Stable Carbenes. Chem. Rev. 2000, 100, 39–92. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.J.; Robertson, K.N.; Masuda, J.D.; Clyburne, J.A. NHC Complexes of Main Group Elements: Novel Structures, Reactivity, and Catalytic Behavior; Wiley-VCH: Weinheim, Germany, 2014; p. 427. [Google Scholar]
- Arduengo, A.J., III; Calabrese, J.; Davidson, F.; Rasika Dias, H.; Goerlich, J.R.; Krafczyk, R.; Marshall, W.J.; Tamm, M.; Schmutzler, R. C−H Insertion reactions of nucleophilic carbenes. Helv. Chim. Acta 1999, 82, 2348–2364. [Google Scholar] [CrossRef]
- Wanzlick, H.W.; Schikora, E. Ein nucleophiles carben. Chem. Ber. 1961, 94, 2389–2393. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Goossen, L.J.; Spiegler, M. Chiral oxazoline/imidazoline-2-ylidene complexes. Organometallics 1998, 17, 2162–2168. [Google Scholar] [CrossRef]
- Sanford, M.S.; Ulman, M.; Grubbs, R.H. New insights into the mechanism of ruthenium-catalyzed olefin metathesis reactions. J. Am. Chem. Soc. 2001, 123, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.A. N-Heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Choi, T.; Grubbs, R.H. Tandem ring-closing metathesis reaction with a ruthenium catalyst containing a N-heterocyclic ligand. Chem. Commun. 2001, 24, 2648–2649. [Google Scholar] [CrossRef]
- Huang, J.; Schanz, H.; Stevens, E.D.; Nolan, S.P. Stereoelectronic effects characterizing nucleophilic carbene ligands bound to the Cp*RuCl (Cp* = η5-C5Me5) moiety: A structural and thermochemical investigation. Organometallics 1999, 18, 2370–2375. [Google Scholar] [CrossRef]
- Breslow, R. On the mechanism of thiamine action. IV. 1 Evidence from studies on model systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Stetter, H. Catalyzed addition of aldehydes to activated double bonds—A new synthetic approach. Angew. Chem. Int. Ed. 1976, 15, 639–647. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Teles, J.H. A novel asymmetric benzoin reaction catalyzed by a chiral triazolium salt. Preliminary communication. Helv. Chim. Acta 1996, 79, 1217–1221. [Google Scholar] [CrossRef]
- Henrique Teles, J.; Melder, J.; Ebel, K.; Schneider, R.; Gehrer, E.; Harder, W.; Brode, S.; Enders, D.; Breuer, K.; Raabe, G. The chemistry of stable carbenes. Part 2. Benzoin-type condensations of formaldehyde catalyzed by stable carbenes. Helv. Chim. Acta 1996, 79, 61–83. [Google Scholar] [CrossRef]
- Kerr, M.S.; Read de Alaniz, J.; Rovis, T. A highly enantioselective catalytic intramolecular stetter reaction. J. Am. Chem. Soc. 2002, 124, 10298–10299. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Connor, E.F.; Nyce, G.W.; Myers, M.; Möck, A.; Hedrick, J.L. First example of N-heterocyclic carbenes as catalysts for living polymerization: Organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002, 124, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Nyce, G.W.; Glauser, T.; Connor, E.F.; Möck, A.; Waymouth, R.M.; Hedrick, J.L. In situ generation of carbenes: A general and versatile platform for organocatalytic living polymerization. J. Am. Chem. Soc. 2003, 125, 3046–3056. [Google Scholar] [CrossRef] [PubMed]
- Grasa, G.A.; Güveli, T.; Singh, R.; Nolan, S.P. Efficient transesterification/acylation reactions mediated by N-heterocyclic carbene catalysts. J. Org. Chem. 2003, 68, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, J.L.; Magbitang, T.; Connor, E.F.; Glauser, T.; Volksen, W.; Hawker, C.J.; Lee, V.Y.; Miller, R.D. Application of complex macromolecular architectures for advanced microelectronic materials. Chem. Eur. J. 2002, 8, 3308–3319. [Google Scholar] [CrossRef]
- Nyce, G.W.; Csihony, S.; Waymouth, R.M.; Hedrick, J.L. A General and Versatile Approach to Thermally Generated N-Heterocyclic Carbenes. Chem. Eur. J. 2004, 10, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The cambridge structural database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Giffin, N.A.; Hendsbee, A.D.; Masuda, J.D. 1,3-Bis(2,6-diisopropylphenyl)imidazolidin-2-ylidene. Acta Crystallogr. 2010, E66, o2194. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- APEX 3, v2016.1-0; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SAINT, v. 8. 37A; Bruker AXS Inc.: Madison, WI, USA, 2015.
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009.





{kind=link}
{kind=link}
{kind=link}
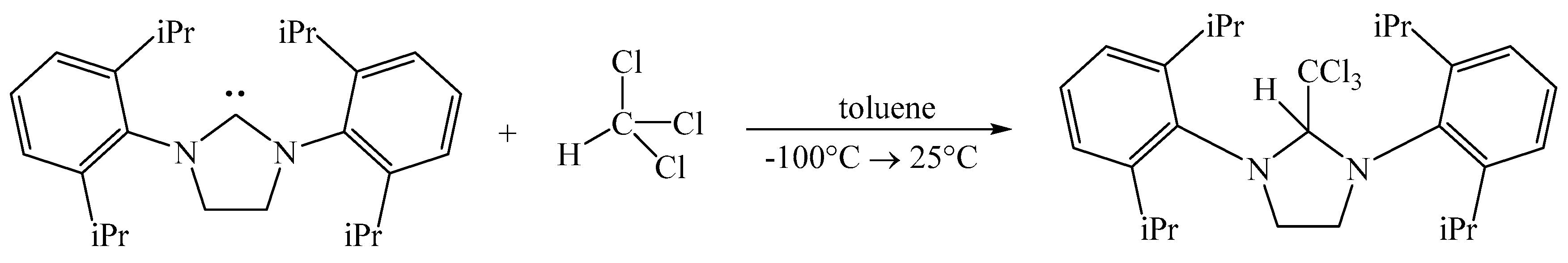{kind=link}
| Bond or Angle Type | Crystal Structure | DFT (B3LYP/6-311G(2d,p)) | ||
|---|---|---|---|---|
| Exp. Bond Length (Å) or Angle (°) | Avg. Bond Length (Å) or Angle (°) | Calc. Bond Length (Å) or Angle (°) | Difference (%) | |
| C–Cl | 1.770(2) | 1.777(3) | 1.8049 | 1.5 |
| 1.7800(19) | ||||
| 1.780(2) | ||||
| Ccarbene–N | 1.442(2) | 1.447(4) | 1.4549 | 0.5 |
| 1.450(3)) | ||||
| Cbackbone–N | 1.464(2) | 1.463(4) | 1.4647 | 0.1 |
| 1.462(3) | ||||
| Ccarbene–C | 1.576(3) | – | 1.5978 | 1.4 |
| N–C–N | 103.23(15) | – | 103.73 | 0.5 |
| C–N–C | 110.26(15) | 110.81(2) | 110.45 | 0.3 |
| 111.36(15) | ||||
| N–Ccarbene–C | 111.22(15) | 112.61(2) | 112.81 | 0.2 |
| 113.99(16) | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stack, D.L.; Masuda, J.D. 1,3-Bis(2,6-diisopropylphenyl)-2-trichloromethylimidazolidine. Molbank 2017, 2017, M962. https://doi.org/10.3390/M962
Stack DL, Masuda JD. 1,3-Bis(2,6-diisopropylphenyl)-2-trichloromethylimidazolidine. Molbank. 2017; 2017(4):M962. https://doi.org/10.3390/M962
Chicago/Turabian StyleStack, Darcie L., and Jason D. Masuda. 2017. "1,3-Bis(2,6-diisopropylphenyl)-2-trichloromethylimidazolidine" Molbank 2017, no. 4: M962. https://doi.org/10.3390/M962
APA StyleStack, D. L., & Masuda, J. D. (2017). 1,3-Bis(2,6-diisopropylphenyl)-2-trichloromethylimidazolidine. Molbank, 2017(4), M962. https://doi.org/10.3390/M962