Synthesis of All Stereoisomers of 1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-N-methyl-1H-pyrido[3,4-b]indole-3-carboxamide


Abstract
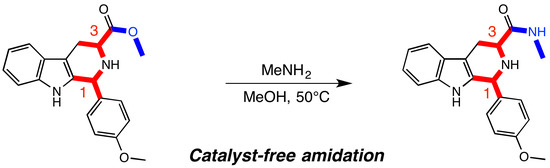
:
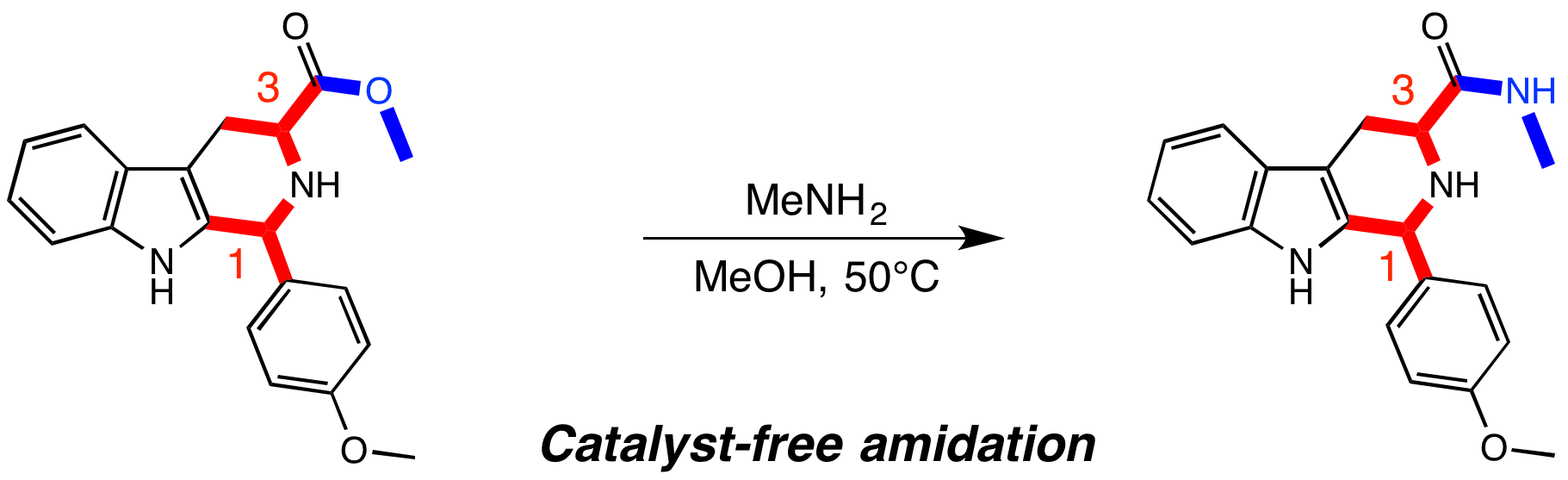
1. Introduction
2. Results
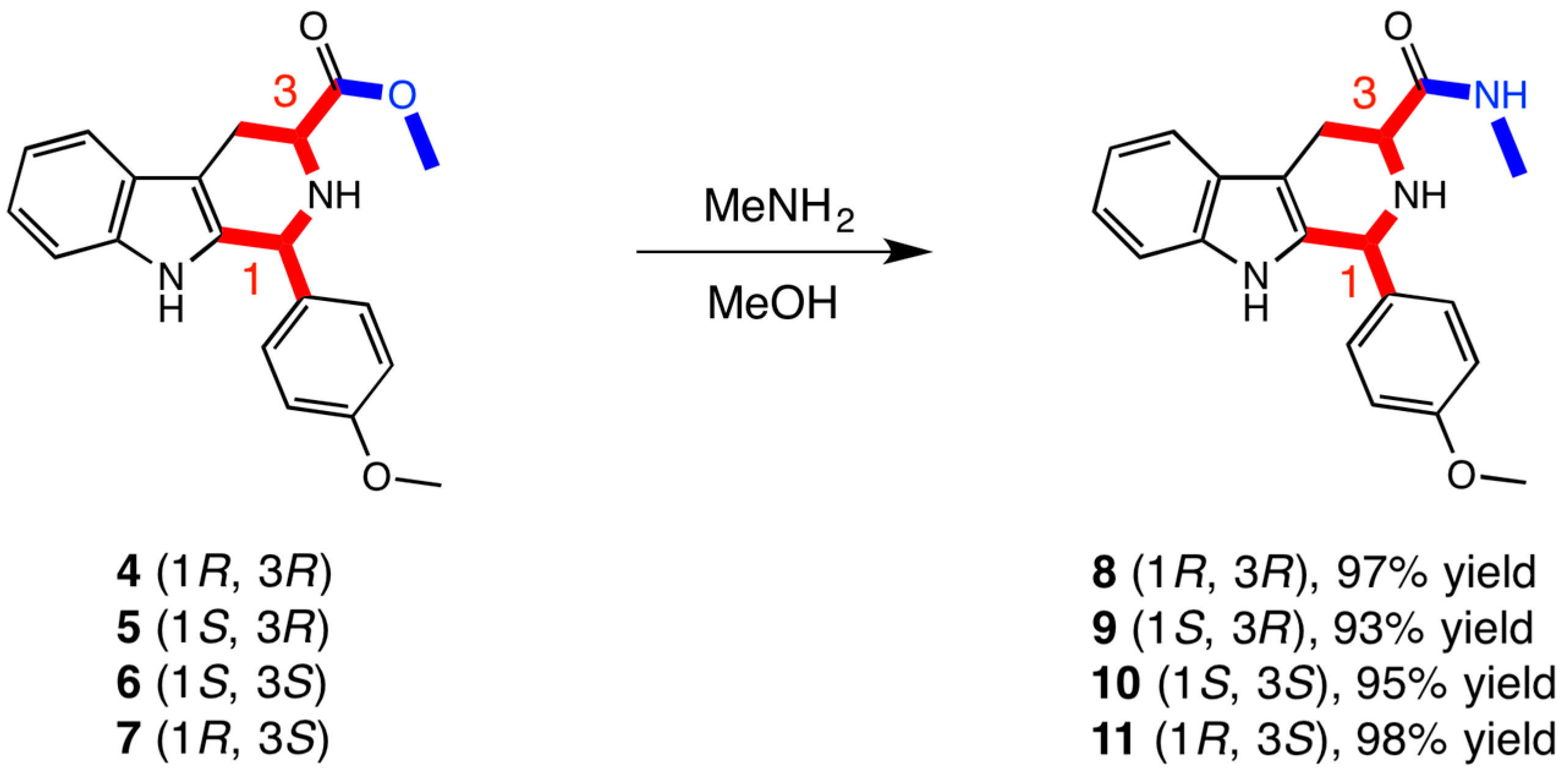
2.1. Chemistry
2.2. Assessment of Cell Viability Using the WST-8 Method
3. Materials and Methods
3.1. Chemistry
3.2. Assessment of Cell Viability Using the WST-8 Method
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rook, Y.; Schmidtke, K.U.; Gaube, F.; Schepmann, D.; Wünsch, B.; Heilmann, J. Bivalent β-carbolines as potential multitarget anti-alzheimer agents. J. Med. Chem. 2010, 53, 3611. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.S.; Koskinen, A.M.P. Harmicine, a tetracyclic tetrahydro-β-carboline: From the first synthetic precedent to isolation from natural sources to target-oriented synthesis (Review). Chem. Heterocycl. Compd. 2015, 50, 1367–1387. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Lazar, C.; Crandall, I.E.; Szarek, W.A. Anti-plasmodium activity of imidazolium and triazolium salts. Bioorg. Med. Chem. 2010, 18, 6184–6196. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, C.; Barresi, V.; Berellini, G.; Musumarra, G. Design and synthesis of trans, 2-(furan-2-yl)vinyl heteroaromatic iodides with antitumour activity. Bioorg. Med. Chem. 2008, 16, 4150. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.K.; He, S.; Guo, L.; Truong, Q.; Qi, H.; Du, W.; Lai, Z.; Liu, J.; Jian, T.; Hong, Q.; et al. Discovery of MK-1421, a potent, selective sstr3 antagonist, as a development candidate for type2 diabetes. ACS Med. Chem. Lett. 2015, 18, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Song, H.J.; Liu, Y.X.; Liu, Y.X.; Huang, Y.Q.; Li, Y.Q.; Wang, Q.M. Design, synthesis, anti-TMV, fungicidal, and insecticidal activity evaluation of 1,2,3,4-tetrahydro-β-carboline-3-carboxylic acid derivatives based on virus inhibitors of plant sources. Bioorg. Med. Chem. Lett. 2014, 24, 5228–5233. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, T.; Galisteo, J. Tetrahydro-β-carboline alkaloids occur in fruits and fruit juices. Activity as antioxidants and radical scavengers. J. Agric. Food Chem. 2003, 51, 7156–7161. [Google Scholar] [CrossRef] [PubMed]
- Price, K.E.; Larrivee-Aboussafy, C.; Lillie, B.M.; McLaughlin, R.W.; Mustakis, J.; Hettenbach, K.W.; Hawkins, J.M.; Vaidyanathan, R. Mild and efficient DBU-catalyzed amidation of cyanoacetates. Org. Lett. 2009, 11, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Fujiwara, R.; Shimizu, Y.; Morisaki, K.; Ohshima, T. Lanthanum (III) triflate catalyzed direct amidation of esters. Org. Lett. 2014, 16, 2018–2021. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Xin, H.L.; Morimoto, H.; Ohshima, T. Direct catalytic alcoholysis of unactivated 8-aminoquinoline amides. ACS Catal. 2017, 7, 3157–3161. [Google Scholar] [CrossRef]
- Muthukrishnan, M.; More, S.V.; Garud, D.R.; Ramana, C.V.; Joshi, R.R.; Joshi, R.A. Pictet-spengler cyclization in room temperature ionic liquid: A convenient access to tetrahydro β-carbolines. J. Heterocycl. Chem. 2006, 43, 767–772. [Google Scholar] [CrossRef]
- Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Water as an efficient medium for the synthesis of tetrahydro-β-carbolines via Pictet–Spengler reactions. Tetrahedron Lett. 2007, 48, 1379–1383. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Methyl Ester Derivatives | Methyl Amide Derivatives | |||||||
|---|---|---|---|---|---|---|---|---|
| 4 (1R,3R) | 5 (1S,3R) | 6 (1S,3S) | 7 (1R,3S) | 8 (1R,3R) | 9 (1S,3R) | 10 (1S,3S) | 11 (1R,3S) | |
| % cell viability | 67.8 | 89.9 | 55.8 | 98.2 | 52.8 | 83.8 | 40.7 | 66.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onda, M.; Higashida, A.; Hirano, T.; Nishio, T.; Hakamata, W. Synthesis of All Stereoisomers of 1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-N-methyl-1H-pyrido[3,4-b]indole-3-carboxamide. Molbank 2018, 2018, M973. https://doi.org/10.3390/M973
Onda M, Higashida A, Hirano T, Nishio T, Hakamata W. Synthesis of All Stereoisomers of 1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-N-methyl-1H-pyrido[3,4-b]indole-3-carboxamide. Molbank. 2018; 2018(1):M973. https://doi.org/10.3390/M973
Chicago/Turabian StyleOnda, Momoko, Ayaka Higashida, Takako Hirano, Toshiyuki Nishio, and Wataru Hakamata. 2018. "Synthesis of All Stereoisomers of 1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-N-methyl-1H-pyrido[3,4-b]indole-3-carboxamide" Molbank 2018, no. 1: M973. https://doi.org/10.3390/M973
APA StyleOnda, M., Higashida, A., Hirano, T., Nishio, T., & Hakamata, W. (2018). Synthesis of All Stereoisomers of 1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-N-methyl-1H-pyrido[3,4-b]indole-3-carboxamide. Molbank, 2018(1), M973. https://doi.org/10.3390/M973