Abstract
1-(2-Hydroxyethyl)imidazolidine-2-thione (1) was obtained as a product from an in situ reaction between N-(2-hydroxyethyl)ethylenediamine, carbon disulfide, potassium hydroxide, and di(4-fluorobenzyl)tin dichloride. Compound 1 was characterized by IR, UV, 1H, 13C{1H}, and 2D (COSY, NOESY, HSQC, and HMBC) NMR spectroscopies. The cyclic molecular structure was confirmed by single crystal X-ray crystallography which showed the five-membered ring to be non-planar and the π-electron density to be localized over the CN2S chromophore. In the crystal, thioamide–N–H…O(hydroxy) and hydroxy–O–H…S(thione) hydrogen bonds lead to supramolecular layers in the bc-plane.
1. Introduction
The title compound, 1-(2-hydroxyethyl)imidazolidine-2-thione (1), as shown in Scheme 1, is in fact a known compound and was probably first described/authenticated in the mid-1950s [1]. Despite there being considerable and long-term chemotherapeutic interest [2,3] in this compound, in more recent times interest has related to its utility in the electroplating of metals [4,5] and as a corrosion inhibitor [6]. Yet, the crystal structure determination has not been described and a detailed spectroscopic analysis is lacking [7]. In the present study, 1 was isolated as a side product from the in situ cyclization reaction of N-(2-hydroxyethyl)ethylenediamine, carbon disulfide, and potassium hydroxide in the presence of di(4-fluorobenzyl)tin dichloride. Such cyclization reactions have been documented in dithiocarbamate chemistry as being a convenient method to form 1-alkyl-2-imidazolidinethiones [8,9], suggesting the organotin reagent is not required for the synthesis. The motivation for the original reaction was to generate bioactive organotin dithiocarbamate compounds inspired by their biological activity, primarily anti-cancer and anti-microbial [10], and as a result of the recent reports of the potential anti-cancer (gold [11], bismuth [12], and zinc [13]) and anti-microbial (copper, silver [14] and gold [15,16]) activities of metal hydroxyethyl-substituted dithiocarbamate compounds, the biological activity of metal dithiocarbamates has been reviewed [17]. Herein, the spectroscopic characterization and X-ray crystal structure determination of 1 are reported.
Scheme 1.
Chemical diagram for 1-(2-hydroxyethyl)imidazolidine-2-thione (1).
2. Results and Discussion
Compound 1 was obtained from an in situ reaction of N-(2-hydroxyethyl)ethylenediamine, carbon disulfide, and potassium hydroxide (normally required to prepare dithiocarbamate anions) with substituted dibenzyltin (IV) chlorides (see Section 3.2); no other compounds were isolated from these reactions. Several closely related cyclic compounds have been observed in the literature as a result of the decomposition reaction between sodium N-isopropyl-N-hydroxyethyldithiocarbamate with a cadmium salt [18], benzimidazole based thiones [19,20], and as a side product from the cyclization of (2-t-butyl)ethanol dithiocarbamate [21]. It is likely the presence of a strong alkaline medium that promotes the formation of these cyclic compounds [22].
IR, UV, 1H, and 13C NMR spectroscopies were employed to characterize 1; see the Supplementary Materials for original spectra. The IR spectrum showed a prominent and broad absorption band due to OH and NH stretching at 3241 cm−1. Other prominent absorption bands were also observed: 1508(s) ν(–NC=S), 1312(m) and 1257(m) ν(C–N), and 1061(s) ν(C–O). In the UV spectrum recorded in methanol solution, a band at 284 nm is assigned to a thione π→π* transition; a similar result was noted in an acetonitrile solution. The assignment of the resonances in the 1H and 13C{1H}-NMR spectra was not straightforward owing to the similarity in the magnetic environments for the four methylene groups. Therefore, 2D NMR (COSY, NOESY, HSQC, and HMBC) experiments were conducted. From the COSY spectrum, a correlation between the OH (δ 2.38) and H5 (δ ~3.87) protons was established, and possibly also between H2 (δ ~3.64) and H3 (δ 3.85). In order to confirm the correlation between the H2 and H3 protons, a NOESY experiment was conducted which indeed confirmed this correlation. Confirmation of the assignments was achieved through HSQC and HMBC experiments, and final assignments are given in Section 3.2.
On the basis of the literature, it is noted that the chemical shifts of O- and N-bound protons can occur virtually anywhere in 1H-NMR spectra. This is because the value does not depend on the exact molecular structure of a compound but is also dependent on the solvent used and the possibility of hydrogen bonding [23,24]. In the 1H spectrum of 1, the chemical shifts for the O- and N-bound protons were shielded and appeared as singlets δ 2.38 and 5.88 ppm, respectively. The relatively high-field shifts for these protons suggest there is only weak or most likely no intramolecular hydrogen bonding in the molecule.
The molecular structure of 1, as determined by single-crystal X-ray crystallography, is shown in Figure 1, and selected geometric parameters are included in the caption. The C1=S1 bond length of 1.6996(13) Å is indicative of a thione bond. The C1–N1 and C1–N2 bond lengths are systematically shorter than the other C–N bonds consistent with some delocalization of π-electron density over the CN2S atoms. The internal C1–N–C angles are both similar and smaller than the C–N2–C4 angles. The same trend is observed for the angles subtended at the C1 atom with the external S1–C1–N angles being approximately 15° wider than the internal N1–C1–N2 angle. The r.m.s. deviation of the five-members comprising the ring is 0.0337 Å with the maximum deviations above and below the least-squares plane being 0.0451(8) and 0.0416(8) Å for the C2 and C3 atoms, respectively, consistent with the ring being twisted around the C2–C3 bond. The S1 (0.0122(19) Å) and C4 (0.025(2) Å) atoms lie to either side of the plane, that is, to the same side as the C2 and C3 atoms, respectively.
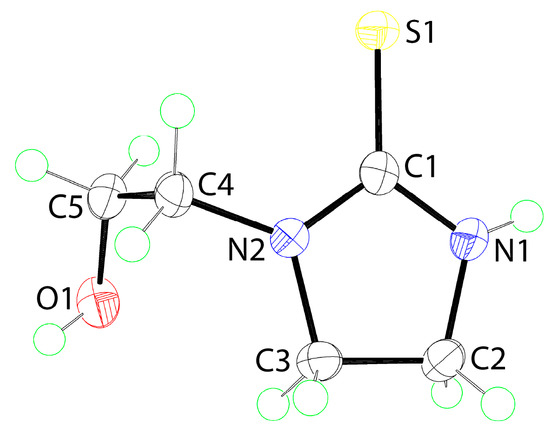
Figure 1.
The molecular structure of 1 showing atom labeling and displacement ellipsoids at the 70% probability level. Selected geometric parameters: C1=S1 = 1.6996(13), C1–O5 = 1.4261(16), C1–N1 = 1.3385(17), C2–N1 = 1.4590(18), C1–N2 = 1.3356(16), C3–N2 = 1.4665(16), C4–N2 = 1.4551(16), and C2–C3 = 1.5342(17) Å; C1–N1–C2 = 112.38(11), C1–N2–C3 = 112.11(10), C1–N2–C4 = 125.49(11), C3–N2–C4 = 122.10(10), S1–C1–N1 = 125.13(10), S1–C1–N2 = 125.33(9), and N1–C1–N2 = 109.53(11)°.
A view of the unit cell contents for 1 is shown in Figure 2a. The most prominent features of molecular packing in the crystal are the formation of thioamide–N–H…O(hydroxy) and hydroxy–O–H…S(thione) hydrogen bonds which combine to form supramolecular layers in the bc-plane (Figure 2b) via the formation of 12-membered {…HO…HNCS}2 synthons which have the form of a chair (Figure 2c); in Graph Set terminology [25], the rings are labeled R44(12). The layers stack along the a-axis without directional interactions between them.
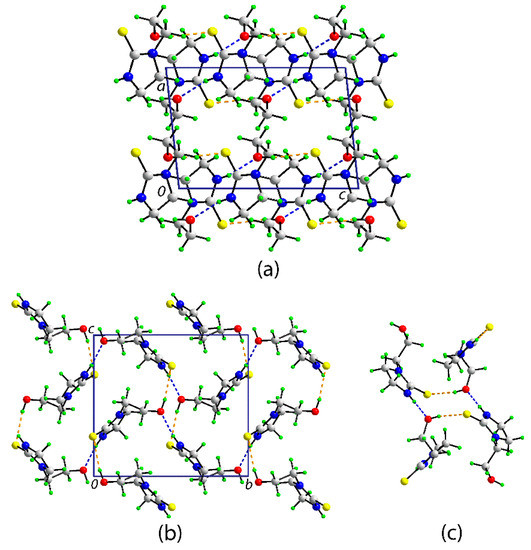
Figure 2.
The molecular packing in the crystal of 1: (a) a view of the unit cell contents in projection down the b-axis, (b) a view of the supramolecular layer parallel to (0 1 1), and (c) detail of the 12-membered {…OH…SCNH}2 synthon. The thioamide–N–H…O(hydroxyl) and hydroxyl–O–H…S(thione) hydrogen bonds are shown as blue and orange dashed lines, respectively. Selected geometric parameters for the specified intermolecular interactions: N1-H1n…O1i = 2.045(17) Å, N1…O1i = 2.8428(15) Å with angle at H1n = 163.8(14)°; O1-H1o…S1ii = 2.324(19) Å, O1…S1ii = 3.1757(10) Å with angle at H1o = 168.2(19)°. Symmetry operations: i 2 − x, −½ + y, ½ – z and ii x, ½ – y, −½ + z.
The most closely related structure to 1 in the literature is one with a benzene ring fused across the C2–C3 bond which, with a bond length of 1.387(3) Å, now has a significant double bond character [18]. The C1–S1, C1–N1, and C1–N2 bond lengths are 1.6801(18), 1.356(3), and 1.362(2) Å, respectively, with the crucial C1–N bond lengths being marginally longer than the equivalent bonds in 1, a result consistent with the significant shortening of the C2–N1 and C3–N2 bond lengths, that is, to 1.379(2) and 1.398(2) Å, compared with 1, and substantial delocalization of π-electron density over the entire ring which is strictly planar [r.m.s. deviation = 0.009 Å].
As intimated above, imidazolidine-2-thiones form an important class of compound and with derivatization possible at both nitrogen atoms and in the ethylene link, it is therefore not surprising there is considerable X-ray crystallographic data available for these derivatives. Possibly, the simplest di-nitrogen substituted compound with an ethylene link is the N,N′-dimethyl-2-imidazolidinethione compound (2) [26]. The substituents at nitrogen can be large and chiral as in the recently report of the (R)-5,6,7,8-tetrahydro-[1,2′-binaphthalen]-8-yl) di-nitrogen substituted derivative (3) [27], again with an ethylene bridge. Substitution can also occur in the ethylene bridge, and related to 1, in the hydroxyethyl chain, with phenyl groups, as in the case of 1-(2-hydroxy-1-phenylethyl)-3-methyl-4-phenylimidazolidine-2-thione (4), implying the formation of two chiral centers [28]. Examples of substantial substitution at all positions around the ring are also exemplified by (E)-3-n-butyl-4-butylidene-1′-methyl-1-phenyl-2-thioxo-1′H-spiro[imidazolidine-5,3′-indol]-2′-one (5) [29] in which one ethylene-carbon is di-substituted, being incorporated in a spiro link, and the other participates in a double bond to an exocyclic carbon atom. The C=S bond lengths in 2, 3, and 5 span the range 1.659(3) Å (5) to 1.684(3) Å (3) with the longer bonds in 1 and 4 (1.697(3) Å) being correlated with their thione sulfur atoms participating in significant hydrogen bonds in their respective crystals. As for 1, the C1–N1, N2 bond lengths are experimentally equivalent in each of 2 (1.342(5) and 1.334(5) Å), 3 (1.361(4) and 1.338(4) Å), 4 (1.345(4) and 1.348(4) Å), and 5 (1.354(4) and 1.358(3) Å).
In conclusion, this study enabled the assignment of the 1H and 13C-NMR resonances for 1 through an analysis of 2D NMR spectra. X-ray crystallography established the non-planar nature of the five-membered ring and π-electron density localized over the CN2S chromophore. In the crystal, supramolecular layers are formed, being sustained by thioamide–N–H…O(hydroxy) and hydroxy–O–H…S(thione) hydrogen bonds.
3. Materials and Methods
3.1. General Information
All chemicals were purchased from commercial sources: Potassium hydroxide, tin powder (Merck, Darmstadt, Germany), 4-fluorobenzyl chloride (Sigma-Aldrich, St. Louis, MI, USA), N-(2-Hydroxyethyl)ethylenediamine (Alfa Aesar, Haverhill, MA, USA), and carbon disulfide (Pancreac, Barcelona, Spain) and used as received with further purification. The solvents used were of EMSURE grade (Merck, Darmstadt, Germany) and used without further purification. The melting point was determined using a Biobase automatic melting point apparatus (Biobase Group, Jinan, Shandong, China) and was uncorrected. The IR spectrum was measured on a Bruker Vertex 70v FTIR (Billerica, MA, USA) spectrophotometer from 4000 to 400 cm−1. Elemental analyses were performed on a Leco TruSpec Micro CHN Elemental Analyzer (Leco, Saint Joseph, MI, USA). The optical absorption spectra were obtained from methanol and acetonitrile solutions of 1 (2 × 10−4 M) in the range 200–800 nm on a Shimadzu UV-3600 plus UV/VIS/NIR spectrophotometer (Shimadzu Corporation, Kyoto Prefecture, Japan). 1H, 13C{1H}, and 2D-NMR experiments (COSY, NOESY, HSQC, and HMBC) were recorded on a Bruker Ascend 400 MHz NMR (Billerica, MA, USA) spectrometer. Compound 1 was dissolved in deuterated chloroform and the chemical shifts were recorded in ppm relative to tetramethylsilane.
3.2. Synthesis and Characterization
Carbon disulfide (0.12 mL, 2 mmol) was slowly added to a stirred solution of N-(2-hydroxyethyl)ethylenediamine (0.20 mL, 2 mmol) in acetone at 0 °C. The solution was stirred for 30 min. Next, potassium hydroxide (50% w/v, 0.23 mL) was added dropwise into the solution which was stirred for another 30 min. Next, di(4-fluorobenzyl)tin dichloride (0.41 g, 1 mmol) in acetone (10 mL) was added into the mixture and stirring was continued for 3 h. The solvent was gradually removed by evaporation until a light brown solid was obtained. The precipitate was recrystallized from acetone to yield pale orange crystals. Similar crystals were isolated when di(4-fluorobenzyl)tin dichloride was substituted with di(4-methylbenzyl)tin dichloride and di(4-chlorobenzyl)tin dichloride. M.p.: 122–124 °C cf. 136.5–137.5 °C [1]. IR (ATR, cm−1) 3241(b) ν(OH, NH), 1508(s) ν(–NC=S), 1480(m) ν(C–C), 1312(m) ν(C–N), 1257(m) ν(C–N), 1061(s) ν(C–O), 932(m) ν(C–C). UV: (methanol): λabs = 284 nm, ε = 5585 L·cm−1·mol−1 and (acetonitrile): λabs = 283 nm, ε = 5850 L·cm−1·mol−1. 1H-NMR (refer to Figure 1 for atom labels): δ 3.88 (H5, 3JHH = 5.08 Hz, 3JH-OH = 4.60 Hz), 3.85 (H3, 3JHH = 9.64 Hz, 4JH-NH = 6.76 Hz), 3.81 (H4, 3JHH = 5.48 Hz, 4JH-OH = 5.20 Hz), 3.63 (H2, 3JHH = 9.52 Hz, 3JH-NH = 8.52 Hz). 13C{1H}-NMR: δ 184.34 (C1), 61.23 (C5), 49.97 (C3), 49.21 (C4), 41.68 (C2).
3.3. X-ray Crystallography
Intensity data for 1 were measured at T = 100(2) K on an Agilent Technologies SuperNova Dual AtlasS2 diffractometer (Rigaku, The Woodlands, TX, USA) fitted with Cu Kα radiation (λ = 1.54184 Å) so that θmax was 67.1°. Data reduction, including absorption correction, was performed with CrysAlis Pro [30]. Of the 8573 reflections measured, 1236 were unique (Rint = 0.025), and of these, 1181 data satisfied the I ≥ 2σ(I) criterion of observability. The structure was solved by direct methods [31] and refined (anisotropic displacement parameters and C-bound H atoms in the riding model approximation) on F2 [32]. The O- and N-bound H atoms were refined without restraint. A weighting scheme of the form w = 1/[σ2(Fo2) + (0.039P)2 + 0.272P] was introduced, where P = (Fo2 + 2Fc2)/3. On the basis of the refinement of 90 parameters, the final values of R and wR (all data) were 0.024 and 0.066, respectively. The molecular structure diagram was generated with ORTEP for Windows [33] and the packing diagrams using DIAMOND [34].
Crystal data for C5H10N2OS (1): M = 146.21, monoclinic, P21/c, a = 6.99980(10), b = 10.3937(2), c = 9.5692(2) Å, β = 95.764(2)°, V = 692.68(2) Å3, Z = 4, Dx = 1.402 g cm−3 and μ = 3.509 mm−1. CCDC deposition number: 1876988.
Supplementary Materials
The following are available online. IR, UV, 1H, 13C{1H}, and 2D (COSY, NOESY, HSQC, and HMBC) NMR spectra, and crystallographic data for 1 in Crystallographic Information File (CIF) format. CCDC 1876988 also contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Author Contributions
S.M.L. was the primary experimentalist, A.H.S.A. performed the NMR experiments, and together they analyzed all data, apart from the X-ray crystallography, which was performed by E.R.T.T.
Funding
This research received no external funding. The APC was funded by Sunway University.
Acknowledgments
The X-ray crystallography laboratory at Sunway University is thanked for providing the X-ray intensity data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McKay, A.F.; Vavasour, G.R. Cyclic thioureas. Can. J. Chem. 1954, 32, 59–62. [Google Scholar] [CrossRef]
- Michels, J.G. 1-(2’-hydroxyethyl)-3-(5-nitrofurfurylideneamino) imidazolidine-2-thione. U.S. Patent DE 1117591, 23 November 1961. [Google Scholar]
- Michels, J.G. 1-(2-hydroxyethyl)-3-amino-2-imidazolidine-thione. U.S. Patent 3,115,499, 24 December 1963. [Google Scholar]
- Qin, Y.; Flajslik, K.; Lefebvre, M. Indium Electroplating Compositions Containing 2-imidazolidinethione Compounds. U.S. Patent 20180016690, 18 January 2018. [Google Scholar]
- Doubina, N.V.; Rigsby, M.A.; Reid, J.D. Chemistry Additives and Process for Cobalt Film Electrodeposition. U.S. Patent 20160273117, 22 September 2016. [Google Scholar]
- Cui, Z.; Duan, Y.; Shen, M.; Li, C.; Liu, X.; Tang, Y. Method for preparing corrosion inhibitor for inhibiting H2S/CO2 corrosion. CN 102268677 A, 7 December 2011. [Google Scholar]
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/1-_2-hydroxyethyl_imidazolidine-2-thione#section=Top (accessed on 21 November 2018).
- Thorn, G.D. 1-Alkyl-2-imidazolidinethiones. Can. J. Chem. 1955, 33, 1278–1279. [Google Scholar] [CrossRef]
- Lo, C.-P. Some 1-alkyl-2-imidazolidinethiones. J. Am. Chem. Soc. 1955, 24, 6667. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Tin dithiocarbamates: Applications and structures. Appl. Organomet. Chem. 2008, 22, 533–550. [Google Scholar] [CrossRef]
- Jamaludin, N.S.; Goh, Z.-J.; Cheah, Y.K.; Ang, K.-P.; Sim, J.H.; Khoo, C.H.; Fairuz, Z.A.; Halim, S.N.B.A.; Ng, S.W.; Seng, H.-L.; et al. Phosphanegold(I) dithiocarbamates, R3PAu[SC(=S)N(iPr)CH2CH2OH] for R = Ph, Cy and Et: Role of phosphane-bound R substituents upon in vitro cytotoxicity against MCF-7R breast cancer cells and cell death pathways. Eur. J. Med. Chem. 2013, 67, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Ishak, D.H.A.; Ooi, K.K.; Ang, K.P.; Akim, A.M.; Cheah, Y.K.; Nordin, N.; Halim, S.N.B.A.; Seng, H.-L.; Tiekink, E.R.T. A bismuth diethyldithiocarbamate compound promotes apoptosis in HepG2 carcinoma, cell cycle arrest and inhibits cell invasion through modulation of the NF-κB activation pathway. J. Inorg. Biochem. 2014, 130, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Ooi, K.K.; Ang, K.P.; Akim, A.M.; Cheah, Y.-K.; Halim, S.N.A.; Seng, H.-L.; Tiekink, E.R.T. Molecular mechanisms of apoptosis and cell selectivity of zinc dithiocarbamates functionalized with hydroxyethyl substituents. J. Inorg. Biochem. 2015, 150, 48–62. [Google Scholar] [CrossRef]
- Jamaludin, N.S.; Halim, S.N.A.; Khoo, C.-H.; Chen, B.-J.; See, T.-H.; Sim, J.-H.; Cheah, Y.-K.; Seng, H.-L.; Tiekink, E.R.T. Bis(phosphane)copper(I) and silver(I) dithiocarbamates: Crystallography and anti-microbial assay. Z. Kristallogr. 2016, 231, 341–349. [Google Scholar] [CrossRef]
- Sim, J.-H.; Jamaludin, N.S.; Khoo, C.-H.; Cheah, Y.-K.; Halim, S.N.B.A.; Seng, H.-L.; Tiekink, E.R.T. In vitro antibacterial and time-kill evaluation of phosphanegold(I) dithiocarbamates, R3PAu[S2CN(iPr)CH2CH2OH] for R = Ph, Cy and Et, against a broad range of Gram-positive and Gram-negative bacteria. Gold Bull. 2014, 47, 225–236. [Google Scholar] [CrossRef]
- Chen, B.-J.; Jamaludin, N.S.; Khoo, C.-H.; See, T.-H.; Sim, J.-H.; Cheah, Y.-K.; Halim, S.N.A.; Seng, H.-L.; Tiekink, E.R.T. In vitro antibacterial and time kill evaluation of mononuclear phosphanegold(I) dithiocarbamates. J. Inorg. Biochem. 2016, 163, 68–80. [Google Scholar] [CrossRef]
- Hogarth, G. Metal-dithiocarbamate complexes: Chemistry and biological activity. Mini Rev. Med. Chem. 2012, 12, 1202–1215. [Google Scholar] [CrossRef]
- Tan, Y.S.; Halim, S.N.A.; Tiekink, E.R.T. Crystal structure of 3-(propan-2-yl)-1,3-oxazolidine-2-thione, C6H11NOS. Z. Kristallogr. New Cryst. Struct. 2014, 229, 55–56. [Google Scholar] [CrossRef]
- Karri, R.; Banerjee, M.; Chalana, A.; Jha, K.K.; Roy, G. Activation of the Hg-C bond of methylmercury by [S2]-donor ligands. Inorg. Chem. 2017, 56, 12102–12115. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Karri, R.; Chalana, A.; Rai, R.K.; Rawat, K.S.; Pathak, B.; Roy, G. Protection of endogenous thiols against methylmercury with benzimidazole-based thione by unusual ligand-exchange reactions. Chem. Eur. J. 2017, 23, 5696–5707. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Karri, R.; Chalana, A.; Rai, R.K.; Rawat, K.S.; Pathak, B.; Roy, G. Chemical detoxification of organomercurials. Angew. Chem. Int. Ed. 2015, 54, 9323–9327. [Google Scholar] [CrossRef] [PubMed]
- de Lima, G.M.; Menezes, D.C.; Dos Santos, J.A.F.; Wardell, J.L.; Filgueiras, C.A.L.; Alcântara, A.F.C. Cyclization of N-alkyldithiocarbamates in alkaline media, a counter example of well-known chemistry-an experimental and theoretical study. J. Coord. Chem. 2012, 65, 559–571. [Google Scholar] [CrossRef]
- Foris, A. On NH NMR Chemical Shifts, Part 1. Available online: https://www.researchgate.net/publication/301653644 (accessed on 9 November 2018).
- Foris, A. On Hydrogen Bonding and OH Chemical Shifts. Available online: https://www.researchgate.net/publication/285927747 (accessed on 9 November 2018).
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: Functionality and Graph Set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Chieh, C.; Cheung, S.K. Crystal structure of N,N’-dimethyl-2-imidazolidinethione, a by-product from the reaction of Na2(CH2N(CH3)CS2)2 and HgCl2. Can. J. Chem. 1983, 61, 211–213. [Google Scholar] [CrossRef]
- Liu, C.; Shen, H.-Q.; Chen, M.-W.; Zhou, Y.-G. C2-symmetric hindered “sandwich” chiral N-heterocyclic carbene precursors and their transition metal complexes: Expedient syntheses, structural authentication, and catalytic properties. Organometallics 2018, 37, 3756–3769. [Google Scholar] [CrossRef]
- Faraji, L.; Jadidi, K.; Notash, B. Synthesis of novel chiral bidentate hydroxyalkyl-N-heterocyclic carbene ligands and their application in palladium-catalyzed Mizoroki–Heck couplings and asymmetric addition of diethylzinc to benzaldehyde. Tetrahedron Lett. 2014, 55, 346–350. [Google Scholar] [CrossRef]
- Antonín Klásek, A.; Mrkvička, V.; Lyčka, A.; Ivan Mikšík, I.; Růžička, A. Reaction of 1-substituted 3-aminoquinoline-2,4-diones with isothiocyanates. An easy pathway to generate novel 2-thioxo-1′H-spiro[imidazoline-5,3′-indole]-2,2′-diones. Tetrahedron 2009, 65, 4908–4916. [Google Scholar] [CrossRef]
- Agilent Technologies Inc. Rigaku Oxford Diffraction, CrysAlis PRO; Agilent Technologies Inc.: Santa Clara, CA, USA, 2017. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).