3. Materials and Methods
All reactions were performed under an atmosphere of nitrogen using solvents dried by standard procedures. Reaction progress was monitored by TLC on Polygram SIL G/UV254 silica gel plates from Macherey and Nagel, Germany. Detection of spots was affected by carbonizing with sulfuric acid (5% in EtOH), staining by spraying the plates with an alkaline aqueous solution of potassium permanganate or by inspection of the TLC plates under UV light (254 nm). Preparative flash chromatography was performed on silica gel (0.032–0.063 mm) from Macherey-Nagel, using plastic cartridges from Götec. The flowrate was regulated by a Sykam S1122 solvent delivery system. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Avance 400 spectrometer and calibrated for the solvent signal (1H: CDCl3: δ = 7.26 ppm; acetone-d6: δ = 2.05 ppm; DMSO-d6: δ = 2.50 ppm; 13C: CDCl3: δ =77.16 ppm; acetone-d6: δ = 29.92 ppm; DMSO-d6: δ = 39.52 ppm). All NMR-assignments were proven by 2D-experiments to be correct. ESI-TOF-HRM spectrometry was performed on a Bruker MAXIS 4G spectrometer. Elemental analyses were obtained from a HEKA tech Euro EA 3000 apparatus. Optical rotations were determined with a Perkin-Elmer Polarimeter 341 in a 10 cm cuvette at 20 °C with a wavelength of 589 nm (Na-lamp). Melting points were measured with a Büchi Melting Point M-560 apparatus.
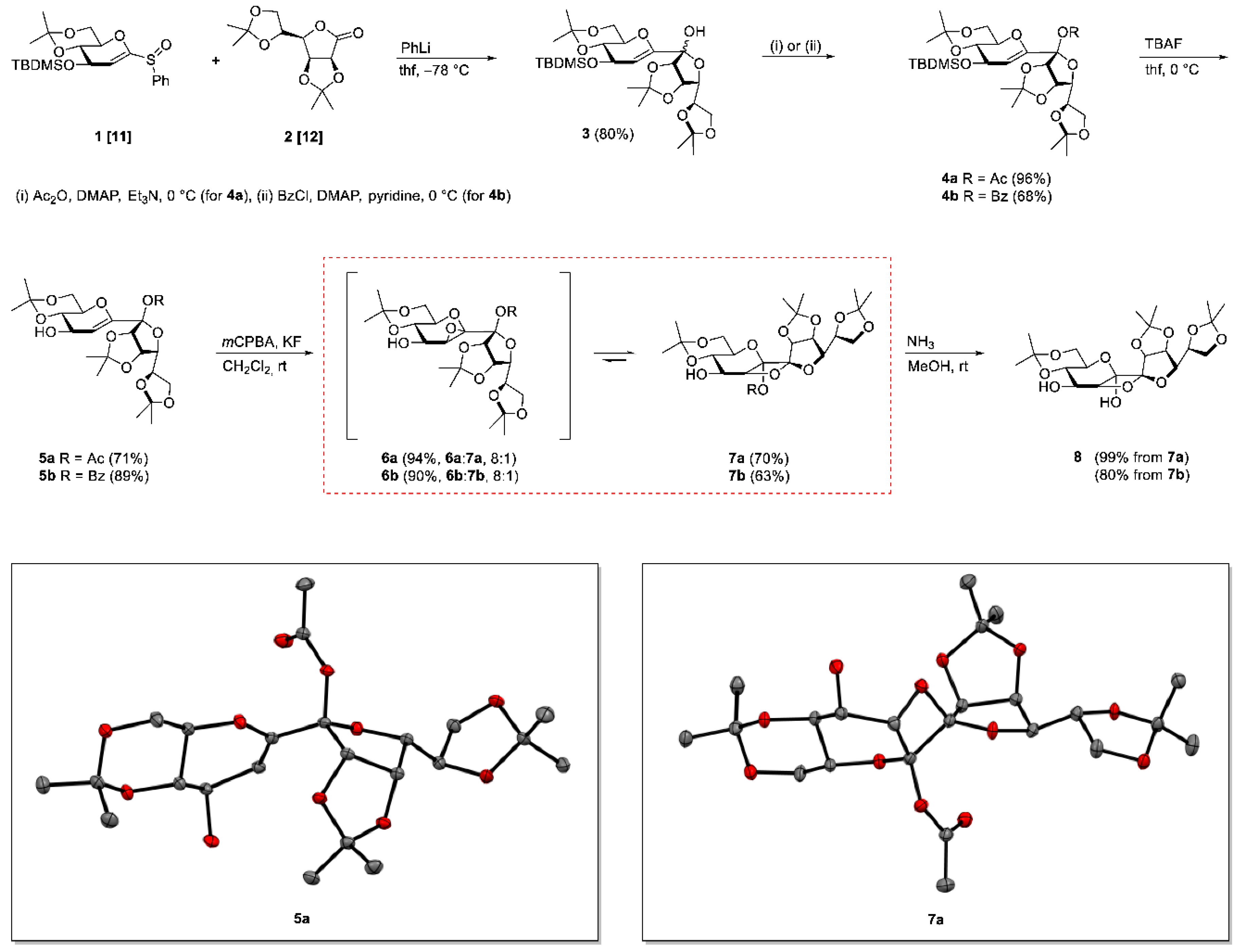
2,6-Anhydro-5-deoxy-4-O-(tert-butyldimethylsilyl)-1,3:8,9:11,12-tri-O-isopropylidene-d-arabino-l-gulo-dodeco-7-ulo-6-enitol (3)
To a suspension of 1 (2.62 g, 6.18 mmol) and grounded molecular sieve (3 Å, 30 mg) in THF (40 mL) was added phenyllithium (1.9 M in Bu2O, 3.57 mL, 6.79 mmol) at −78 °C over a period of 15 min. After 5 min a further solution containing 2 (1.75 g, 6.79 mmol) in THF (20 mL) was added dropwise over a period of 30 min. Subsequently, the reaction mixture was stirred for 2 h at the same temperature before the cooling bath was removed. The reaction was quenched by addition of sat. aq. NH4Cl-solution (60 mL) at ambient temperature and the molecular sieve was filtered off. The organic layer was separated, the aqueous layer was extracted with CH2Cl2 (3 × 20 mL) and the combined organic layers were dried with Na2SO4. Removing of the solvent followed by column chromatography (methylene chloride/ethyl acetate 5:1) furnished 3 (2.77 mg, 4.97 mmol, 80%, anomeric mixture α:β, 1:5) as a colorless crystalline solid. Rf = 0.22–0.46 (methylene chloride/ethyl acetate 5:1).
3β*(major anomer): 1H-NMR (400 MHz, CDCl3): δ(ppm) = 5.04 (d, J5,4 = 2.2 Hz, 1H, H-5), 4.82 (dd, J9,8 = 5.7 Hz, J9,10 = 3.5 Hz, 1H, H-9), 4.53 (d, J8,9 = 5.9 Hz, 1H, H-8), 4.38–4.44 (m, 1H, H-11), 4.33–4.35 (m, 1H, H-4), 4.15 (dd, J10,11 = 7.2 Hz, J10,9 = 3.5 Hz, 1H, H-10), 4.07–4.11 (m, 1H, H-12a), 4.03 (dd, J = 8.7 Hz, J = 4.9 Hz, 1H, H-12b), 3.86–3.95 (m, 2H, H-1a, H-1b), 3.71–3.76 (m, 2H, H-2, H-3), 2.77 (s, 1H, OH), 1.50, 1.44, 1.42, 1.40, 1.38, 1.31 (6s, 18H, C(CH3)2), 0.87 (s, 9H, SiC(CH3)3), 0.07, 0.06 (2s, 6H,SiCH3). 13C-NMR (101 MHz, CDCl3): δ(ppm) = 150.4 (C-6), 113.3, 109.1 (C(CH3)2), 104.3 (C-5), 103.4 (C-7), 99.4 (C(CH3)2), 86.7 (C-8), 80.0 (C-9), 79.4 (C-10), 73.2 (C-11), 72.9 (C-3), 70.2 (C-2), 68.2 (C-4), 66.7 (C-12), 61.7 (C-1), 28.9, 26.8, 25.8 (C(CH3)2), 25.7 (SiC(CH3)3), 25.4, 25.0, 19.1 (C(CH3)2), 18.1 (SiC(CH3)3), –4.4, –4.8 (SiCH3).
3α*(minor anomer): 1H-NMR (400 MHz, CDCl3): δ(ppm) = 5.07 (d, J5,4 = 2.1 Hz, 1H, H-5), 4.79 (dd, J9,8 = 6.0 Hz, J9,10 = 3.7 Hz, 1H, H-9), 4.62 (d, J8,9 = 6.0 Hz, 1H, H-8), 4.34–4.35 (m, 2H, H-4, H-11), 4.10–4.11 (m, 1H, H-12a), 4.01–4.02 (m, 1H, H-12b), 3.85–3.88 (m, 2H, H-1a, H-1b, 3.76–3.80 (m, 3H, H-2, H-3, H-10), 2.77 (s, 1H, OH), 1.57, 1.49, 1.42, 1.40, 1.36 (5s, 18H, C(CH3)2), 0.88 (s, 9H, SiC(CH3)3), 0.08, 0.07 (2s, 6H, SiCH3). 13C-NMR (101 MHz, CDCl3): δ(ppm) = 150.8 (C-6), 113.6, 109.3 (C(CH3)2), 103.1 (C-5), 100.6 (C-7), 99.6 (C(CH3)2), 80.7 (C-8), 80.0 (C-9), 78.6 (C-10), 73.2 (C-11), 72.8 (C-3), 70.3 (C-2), 67.8 (C-4), 67.2 (C-12), 61.7 (C-1), 28.9, 26.9, 25.9 (C(CH3)2), 25.8 (SiC(CH3)3), 25.2, 24.6, 19.0 (C(CH3)2), 18.2 (SiC(CH3)3), –4.4, –4.8 (SiCH3). HRESIMS m/z 581.27570 (calcd for C27H46O10SiNa, 581.27524); anal. C 58.37, H 8.21, calcd for C27H46O10Si, C 58.04, H 8.39. *Anomers 3β and 3α could be interchanged
2,6-Anhydro-5-deoxy-7-O-acetyl-4-O-(tert-butyldimethylsilyl)-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-d-lyxo-dodeco-6-enitol (4a)
A solution of 3 (300 mg, 0.537 mmol) and 4-dimethylaminopyridine (13 mg, 0.107 mmol) in triethylamine (7 mL) was treated with acetic anhydride (508 μL, 5.37 mmol) at 0 °C. After stirring for 12 h at ambient temperature until TLC indicated complete transformation of the starting material, the reaction mixture was diluted with ethyl acetate (20 mL). The organic phase was washed with 1 N hydrochloric acid (3 × 10 mL), sat. aq. NH4Cl-solution (1 × 10 mL) and brine (1 × 10 mL). The solution was dried with Na2SO4, the solvent was removed and the crude product was purified by column chromatography (petroleum ether/ethyl acetate 5:1) to afford 4a (309 mg, 0.515 mmol, 96%) as a colorless crystalline solid. Rf = 0.82 (methylene chloride/ethyl acetate 5:1). = +64.3 (c = 1.0, CHCl3). M.p. 52 °C (n-hexane). 1H-NMR (400 MHz, CDCl3): δ (ppm) = 4.99 (d, J5,4 = 2.2 Hz, 1H, H-5), 4.86 (dd, J9,8 = 5.6 Hz, J9,10 = 3.4 Hz, 1H, H-9), 4.69 (d, J8,9 = 5.6 Hz, 1H, H-8), 4.42 (ddd, J = 8.3 Hz, J = 5.9 Hz, J = 3.9 Hz, 1H, H-11), 4.35 (dd, J4,3 = 6.7 Hz, J4,5 = 2.1 Hz, 1H, H-4), 4.09–4.15 (m, 1H, H-12a), 3.99–4.07 (m, 2H, H-10, H-12b), 3.80–3.89 (m, 2H, H-1aH-1b), 3.61–3.73 (m, 2H, H-2, H-3), 2.01 (s, 3H, COCH3), 1.48, 1.47, 1.45, 1.37, 1.37, 1.33 (6s, 18H, C(CH3)2), 0.83 (s, 9H, SiC(CH3)3), 0.03, 0.03 (2s, 6H, SiCH3).13C-NMR (101 MHz, CDCl3): δ(ppm) = 168.3 (CO), 146.9 (C-6), 113.9, 109.4 (C(CH3)2), 107.4 (C-7), 104.8 (C-5), 99.3 (C(CH3)2), 86.4 (C-8), 81.2 (C-10), 79.6 (C-9), 73.0 (C-3), 72.8 (C-11), 70.2 (C-2), 68.2 (C-4), 67.1 (C-12), 61.7 (C-1), 28.9, 27.0, 25.8 (C(CH3)2), 25.6 (SiC(CH3)3), 25.1, 25.1 (C(CH3)2), 21.5 (COCH3), 19.1 (C(CH3)2), 18.0 (SiC(CH3)3), –4.4, –4.9 (SiCH3). HRESIMS m/z 623.28604 (calcd for C29H48O11SiNa, 623.28581); anal. C 58.10, H 8.21, calcd for C29H48O11Si, C 57.98, H 8.05.
2,6-Anhydro-5-deoxy-7-O-benzoyl-4-O-(tert-butyldimethylsilyl)-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-d-lyxo-dodeco-6-enitol (4b)
A solution of 3 (930 mg, 1.67 mmol) and DMAP (41 mg, 0.333 mmol) in pyridine (15 mL) was treated with benzoyl chloride (1.92 mL, 16.7 mmol) at 0 °C. After stirring for 48 h at ambient temperature TLC indicated complete transformation of the starting material and the reaction mixture was diluted with ethyl acetate (60 mL). The organic layer was washed with 1 N hydrochloric acid (3 × 30 mL), sat. aq. NaHCO3-solution (3 × 20 mL) and brine (1 × 20 mL). The solution was dried with Na2SO4, the solvent was removed and the crude product was purified by column chromatography (toluene/ethyl acetate, gradient: 100:1 – 20:1) to afford 4b (749 mg, 1.13 mmol, 68%) as a colorless crystalline solid. Rf = 0.66 (toluene/ethyl acetate 4:1). = +57.1(c = 1.0, CHCl3). M.p. 58 °C. 1H-NMR (400 MHz, CDCl3): δ(ppm) = 7.90–8.02 (m, 2H, Ph), 7.52–7.62 (m, 1H, Ph), 7.39–7.48 (m, 2H, Ph), 5.11 (d, J5,4 = 2.3 Hz, 1H, H-5), 4.90–5.01 (m, 2H, H-8, H-9), 4.47 (ddd, J = 8.2 Hz, J = 6.0 Hz, J11,12b = 4.0 Hz, 1H, H-11), 4.41 (dd, J4,3 = 6.7 Hz, J4,5 = 2.2 Hz, 1H, H-4), 4.08–4.17 (m, 2H, H-10, H-12a), 4.05 (dd, J12b,12a = 9.1 Hz, J12b,11 = 4.0 Hz, 1H, H-12b), 3.66–3.84 (m, 4H, H-1a, H-1b, H-2, H-3), 1.52, 1.48, 1.42, 1.38, 1.37 (5s, 18H, C(CH3)2), 0.86 (s, 9H, SiC(CH3)3), 0.06, 0.06 (2s, 6H, SiCH3).13C-NMR (101 MHz, CDCl3): δ(ppm) = 164.0 (CO), 147.3 (C-6), 133.4, 130.2, 129.9, 128.5 (Ph), 114.2, 109.6 (C(CH3)2), 108.2 (C-7), 105.1 (C-5), 99.5 (C(CH3)2), 86.7 (C-8), 81.8 (C-10), 80.0 (C-9), 73.3 (C-3), 73.0 (C-11), 70.4 (C-2), 68.5 (C-4), 67.3 (C-12), 61.8 (C-1), 29.1, 27.1, 26.0 (C(CH3)2), 25.8 (SiC(CH3)3), 25.4, 25.3, 19.3 (C(CH3)2), 18.2 (SiC(CH3)3), –4.2, –4.7 (SiCH3). HRESIMS m/z 685.30154 (calcd for C34H50O11SiNa, 685.30146); anal. C 61.26, H 8.02, calcd for C34H50O11Si, C 61.61, H 7.60.
General Procedure A for Desilylation of 4a and 4b
Tetra-n-butylammonium fluoride (1.0 M in THF, 1.5 eq.) was added to a solution of 4a or 4b (1.0 mmol) in THF (10 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirring was continued until complete conversion of the starting material was detected by TLC. Subsequently, the solution was diluted with CH2Cl2 (20 mL) and washed with brine (3 × 10 mL). The aqueous phase was extracted with CH2Cl2 (2 × 20 mL) and the combined organic phases were dried with Na2SO4. The solvent was evaporated and the residue was purified by column chromatography (petroleum ether/ethyl acetate 2:1 containing 0.5% Et3N).
2,6-Anhydro-5-deoxy-7-O-acetyl-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-d-lyxo-dodeco-6-enitol (5a)
Following general procedure A 5a was obtained as a white amorphous solid and was prepared from 4a in 71% yield stirring the reaction mixture for 20 h. Rf = 0.28 (petroleum ether/ethyl acetate 1:1). = +98.0 (c = 1.0, CHCl3). M.p. 179 °C (n-hexane/ethyl acetate). 1H-NMR (400 MHz, CDCl3): δ(ppm) = 5.14 (d, J5,6 = 2.2 Hz, 1H, H-5), 4.88 (dd, J9,8 = 5.7 Hz, J9,10 = 3.5 Hz, 1H, H-9), 4.71 (d, J8,9 = 5.7 Hz, 1H, H-8), 4.37–4.45 (m, 2H, H-4, H-11), 4.01–4.12 (m, 3H, H-10, H-12a, H-12b), 3.85–3.94 (m, 2H, H-1a, H-1b), 3.68–3.80 (m, 2H, H-2, H-3), 2.02 (s, 3H, COCH3), 1.53, 1.47, 1.45, 1.42, 1.37, 1.33 (6s, 18H, C(CH3)2). 13C-NMR (101 MHz, CDCl3): δ (ppm) = 168.2 (CO), 148.0 (C-6), 113.7, 109.4 (C(CH3)2), 107.1 (C-7), 102.9 (C-5), 99.7 (C(CH3)2), 86.3 (C-8), 81.5 (C-10), 79.6 (C-9), 73.2 (C-3), 72.8 (C-11), 69.9 (C-2), 67.8 (C-4), 67.0 (C-12), 61.5 (C-1), 28.9, 27.0, 25.8, 25.1, 24.9 (C(CH3)2), 21.6 (COCH3), 19.2 (C(CH3)2). HRESIMS m/z 509.19928 (calcd for C23H34O11Na, 509.19933); anal. C 56.57, H 7.17, calcd for C23H34O11, C 56.78, H 7.04.
2,6-Anhydro-5-deoxy-7-O-benzoyl-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-d-lyxo-dodeco-6-enitol (5b)
Following general procedure A 5b was obtained as a colorless amorphous solid and was prepared from 4b in 89% yield stirring the reaction mixture for 24 h. Rf = 0.37 (petroleum ether/ethyl acetate 1:1). = +89.4 (c = 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ(ppm) = 7.92–7.99 (m, 2H, Ph), 7.54–7.60 (m, 1H, Ph), 7.41–7.47 (m, 2H, Ph), 5.26 (d, J5,4 = 2.2 Hz, 1H, H-5), 4.99 (dd, J9,8 = 5.9 Hz, J9,10 = 3.6 Hz, 1H, H-9), 4.95 (d, J8,9 = 5.9 Hz, 1H, H-8), 4.41–4.50 (m, 2H, H-4, H-11), 4.09–4.14 (m, 2H, H-10, H-12a), 4.04 (dd, J = 9.1 Hz, J = 4.2 Hz, 1H, H-12b), 3.72–3.88 (m, 4H, H-1a, H-1b, H-2, H-3), 2.11 (br. s., 1H, OH), 1.53, 1.52, 1.42, 1.41, 1.38, 1.36 (6 s, 18H, C(CH3)2).13C-NMR (101 MHz, CDCl3): δ(ppm) = 163.9 (CO), 148.4 (C-6), 133.5, 130.1, 129.9, 128.6 (Ph), 114.0, 109.6 (C(CH3)2), 107.9 (C-7), 103.2 (C-5), 99.9 (C(CH3)2), 86.6 (C-8), 82.0 (C-10), 79.9 (C-9), 73.4 (C-3), 73.0 (C-11), 70.2 (C-2), 68.1 (C-4), 67.2 (C-12), 61.6 (C-1), 29.1, 27.1, 26.0, 25.2, 25.1, 19.4 (C(CH3)2). HRESIMS m/z 571.21543 (calcd for C28H36O11Na, 571.21498); anal. C 61.18, H 6.77, calcd for C28H36O11, C 61.30, H 6.61.
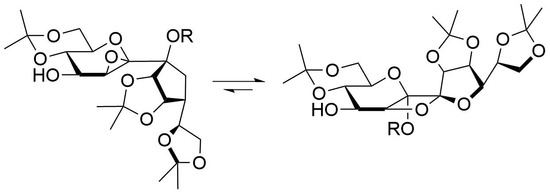
General Procedure B for Epoxidation of 5a and 5b and Further Rearrangement to the Oxetanes
To a solution of mCPBA (2.5 eq.) in CH2Cl2 (10 mL) was added potassium fluoride (5 eq.) and the resulting suspension was stirred for 30 min at ambient temperature. A solution containing 5a or 5b (0.5 mmol) in CH2Cl2 (10 mL) was added in one portion and stirring was continued for 2 h (epoxidation). To enforce the rearrangement to the oxetanes the reaction mixture was stirred for 20 h, the white precipitate was filtered off and the solution was stirred for further 20 h. The reaction was quenched by addition of sat. aq. NaHCO3-solution (10 mL) and sat. aq. Na2S2O3-solution (10 mL). Subsequently, the aqueous layer was extracted by CH2Cl2 (3 × 10 mL), the combined organic layers were dried with Na2SO4, the solvent was removed and the crude product was purified by column chromatography.
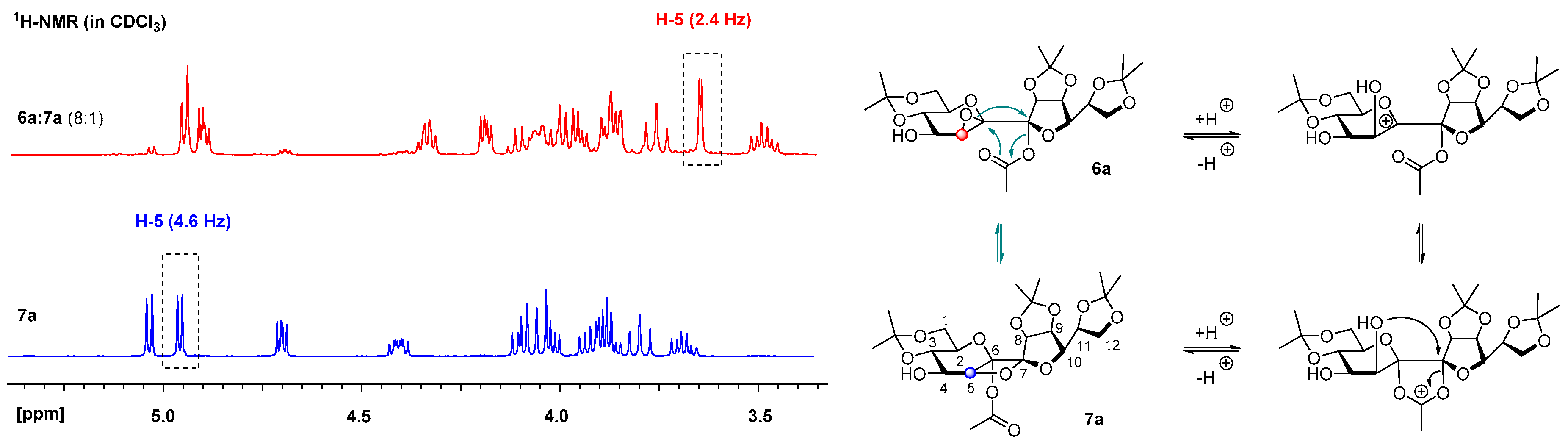
5,6-Anhydro-7-O-acetyl-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-β-d-manno-dodeco-6,7-diulo-2,6-pyranose-7,10-furanose (6a)
Following general procedure B a mixture of 6a and 7a (8:1) was obtained after quenching the reaction after 2 h as a white amorphous solid and was prepared from 5a in 94% yield. A mixture of petroleum ether/ethyl acetate 3:1 containing 0.5% Et3N was used as the eluent for column chromatography. Rf = 0.58 (petroleum ether/ethyl acetate 1:2). 1H-NMR (400 MHz, CDCl3): δ(ppm) = 4.96 (d, J8,9 = 6.0 Hz, 1H, H-8), 4.91 (dd, J9,8 = 6.0 Hz, J9,10 = 3.9 Hz, 1H, H-9), 4.31–4.38 (m, 1H, H-11), 4.20 (dd, J10,11 = 6.6 Hz, J10,9 = 3.9 Hz, 1H, H-10), 3.85–4.06 (m, 5H, H-1a, H-3, H-4, H-12a, H-12b), 3.77 (dd, J = 10.7 Hz, J = 10.7 Hz, 1H, H-1b), 3.66 (d, J5,4 = 2.4 Hz, 1H, H-5), 3.49 (ddd, J = 10.2 Hz, J = 10.2 Hz, J = 5.7 Hz, 1H, H-2), 2.38 (br. s., 1H, OH), 2.06 (s, 3H, COCH3), 1.57, 1.48, 1.42, 1.39, 1.35, 1.33 (6s, 18H, C(CH3)2). 13C-NMR (101 MHz, CDCl3): δ(ppm) = 169.2 (CO), 114.2, 109.2 (C(CH3)2), 107.9 (C-7), 99.7 (C(CH3)2), 86.2 (C-8), 85.0 (C-6), 82.9 (C-10), 79.9 (C-9), 73.2 (C-11), 73.0 (C-3), 70.6 (C-2), 69.9 (C-4), 66.4, 61.7 (C-1), 57.3 (C-5), 29.1, 27.0, 25.6, 25.3, 24.6 (C(CH3)2), 21.9 (COCH3), 19.3 (C(CH3)2). HRESIMS m/z 525.19448 (calcd for C23H34O12Na, 525.19425).
5,6-Anhydro-7-O-benzoyl-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-β-d-manno-dodeco-6,7-diulo-2,6-pyranose-7,10-furanose (6b)
Following general procedure B, a mixture of 6b and 7b (8:1) was obtained after quenching the reaction after 2 h as a white amorphous solid and was prepared from 5b in 90% yield. A mixture of petroleum ether/ethyl acetate 3:1 containing 0.5% Et3N was used as the eluent for column chromatography. Rf = 0.64 (petroleum ether/ethyl acetate 1:2). 1H-NMR (400 MHz, CDCl3): δ(ppm) = 7.92–7.99 (m, 2H, Ph), 7.59–7.63 (m, 1H, Ph), 7.45–7.50 (m, 2H, Ph), 5.18 (d, J8,9 = 6.0 Hz, 1H, H-8), 5.03 (dd, J9,8 = 6.1 Hz, J9,10 = 4.0 Hz, 1H, H-9), 4.34–4.41 (m, 1H, H-11), 4.31 (dd, J10,11 = 6.6 Hz, J10,9 = 3.9 Hz, 1H, H-10), 4.13–4.18 (m, 1H, H-4), 4.03 (dd, J = 8.9 Hz, J = 6.2 Hz, 1H, H-12a), 3.91–3.99 (m, 2H, H-3, 12b), 3.73–3.83 (m, 3H, H-1a, H-1b, H-5), 3.54 (ddd, J = 10.0 Hz, J = 10.0 Hz, J = 6.0 Hz, 1H, H-2), 2.42 (br. s., 1H, OH), 1.62, 1.48, 1.39, 1.37, 1.34 (5s, 18H, C(CH3)2). 13C-NMR (101 MHz, CDCl3): δ(ppm) = 164.7 (CO), 133.8, 129.9, 129.8, 128.8 (Ph), 114.3, 109.2 (C(CH3)2), 108.5 (C-7), 99.7 (C(CH3)2), 86.4 (C-8), 85.3 (C-6), 83.2 (C-10), 80.0 (C-9), 73.2 (C-11), 73.0 (C-3), 70.7 (C-2), 69.9 (C-4), 66.4 (C-12), 61.6 (C-1), 57.4 (C-5), 29.0, 26.9, 25.6, 25.3, 24.6, 19.3 (C(CH3)2). HRESIMS m/z 587.21029 (calcd for C28H36O12Na, 587.20990).
5,7-Anhydro-6-O-acetyl-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-β-d-manno-dodeco-6,7-diulo-2,6-pyranose-7,10-furanose (7a)
Following general procedure B 7a was obtained after quenching the reaction after 40 h as a white crystalline solid and was prepared from 5a in 70% yield. A mixture of petroleum ether/ethyl acetate 2:1 was used as the eluent for column chromatography. Crystals from 7a were grown by overlaying a saturated solution of 7a in methylene chloride with n-heptane and slowly evaporating the methylene chloride. Rf = 0.51 (petroleum ether/ethyl acetate 1:2). = +18.5 (c = 1.0, CHCl3). M.p. 151 °C (n-hexane/ethyl acetate). 1H-NMR (400 MHz, CDCl3): δ(ppm) = 5.04 (d, J8,9 = 5.6 Hz, 1H, H-8), 4.96 (d, J4,5 = 4.9 Hz, 1H, H-5), 4.70 (dd, J9,8 = 5.6 Hz, J9,10 = 4.0 Hz, 1H, H-9), 4.37–4.46 (m, 1H, H-11), 4.00–4.13 (m, 3H, H-3, H-12a, H12b), 3.86–3.97 (m, 3H, H-1a, H-4, H-10), 3.80 (dd, J = 10.5 Hz, 1H, H-1b), 3.65–3.74 (m, 1H, H-2), 2.14 (d, JOH,4 = 9.0 Hz, 1H, OH), 2.10 (s, 3H, COCH3), 1.56, 1.55, 1.43, 1.43, 1.39, 1.36 (6s, 18H, C(CH3)2). 13C-NMR (101 MHz, CDCl3): δ(ppm) = 169.4 (CO), 114.2 (C(CH3)2), 112.9 (C-7), 109.5, 99.9 (C(CH3)2), 98.3 (C-6), 80.9 (C-5), 80.0 (C-10), 79.1 (C-8), 78.6 (C-9), 73.5 (C-11), 71.6 (C-3), 69.1 (C-2), 68.9 (C-4), 67.1 (C-12), 61.8 (C-1), 29.1, 27.1, 25.9, 25.8, 25.4 (C(CH3)2), 21.2 (COCH3), 19.2 (C(CH3)2). HRESIMS m/z 525.19318 (calcd for C23H34O12Na, 525.19425); anal. C 55.08, H 7.11, calcd for C23H34O12, C 54.97, H 6.82.
5,7-Anhydro-6-O-benzoyl-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-β-d-manno-dodeco-6,7-diulo-2,6-pyranose-7,10-furanose (7b)
Following general procedure B 7b was obtained after quenching the reaction after 40 h as a white amorphous solid and was prepared from 5b in 63% yield. A mixture of petroleum ether/ethyl acetate 2:1 was used as the eluent for column chromatography. Rf = 0.60 (petroleum ether/ethyl acetate 1:2). = +15.7 (c = 1.0, CHCl3). 1H-NMR (400 MHz, acetone-d6): δ(ppm) = 8.04–8.10 (m, 2H, Ph), 7.68–7.74 (m, 1H, Ph), 7.53–7.59 (m, 2H, Ph), 5.16 (d, J8,9 = 5.6 Hz, 1H, H-8), 5.10 (d, J5,4 = 4.5 Hz, 1H, H-5), 4.82 (dd, J9,8 = 5.6 Hz, J9,10 = 3.7 Hz, 1H, H-9), 4.36 (ddd, J = 6.5 Hz, J = 6.5 Hz, J = 5.1 Hz, 1H, H-11), 4.15–4.26 (m, 2H, H-3, OH), 3.99–4.06 (m, 3H, H-4, H-10, H-12a), 3.80–3.97 (m, 4H, H-1a, H-1b, H-2, H-12b), 1.52, 1.51, 1.37, 1.35, 1.34, 1.28 (6s, 18H, C(CH3)2). 13C-NMR (101 MHz, acetone-d6): δ(ppm) = 165.7 (CO), 135.0, 130.8, 130.2, 129.7 (Ph), 114.2 (C(CH3)2), 113.5 (C-7), 109.5, 100.2 (C(CH3)2), 99.5 (C-6), 83.3 (C-5), 80.1 (C-10), 79.6 (C-9), 79.6 (C-8), 74.5 (C-11), 72.1 (C-3), 70.7 (C-2), 69.4 (C-4), 67.1 (C-12), 62.4 (C-1), 29.5, 27.2, 26.3, 26.2, 25.7, 19.3 (C(CH3)2). HRESIMS m/z 587.20977 (calcd for C28H36O12Na, 587.20990); anal. C 59.64, H 6.86, calcd for C28H36O12, C 59.57, H 6.43.
5,7-Anhydro-1,3:8,9:11,12-tri-O-isopropylidene-β-d-manno-β-d-manno-dodeco-6,7-diulo-2,6-pyranose-7,10-furanose (8)
From 7a: 7a (50 mg, 0.10 mmol) was dissolved in methanol (2 mL) and ammonia (7 N in methanol, 280 μL, 1.99 mmol) was added at ambient temperature. TLC indicated complete transformation of the starting material after 5 h and the volatile components were removed. Neat 8 (46 mg, 0.099 mmol, 99%) remained as a white crystalline solid.
From 7b: 7b (20 mg, 0.0354 mmol) was dissolved in methanol (1 mL) and ammonia (7 N in methanol, 101 μL, 0.708 mmol) was added at ambient temperature. TLC indicated complete transformation of the starting material after 5 h and the volatile components were removed. Subsequently, the crude product was purified by column chromatography (petroleum ether/ethyl acetate 1:2) and 8 (13 mg, 0.282 mmol, 80%) was isolated as a white crystalline solid.
Rf = 0.32 (petroleum ether/ethyl acetate 1:3). = +17.5 (c = 1.0, CHCl3). M.p. 122 °C (n-hexane). 1H-NMR (600 MHz, DMSO-d6): δ(ppm) = 7.09 (s, 1H, OH-6), 5.16 (d, JOH,4 = 5.7 Hz, 1H, OH-4), 4.75 (d, J8,9 = 5.9 Hz, 1H, H-8), 4.57–4.63 (m, 2H, H-5, H-9), 4.22 (ddd, J11,10 = 7.3 Hz, J11,12a = 6.4 Hz, J11,12b = 5.1, 1H, H-11), 4.01 (dd, J12a,12b = 8.4 Hz, J12a,11 = 6.4 Hz, 1H, H-12a), 3.92 (dd, J12b,12a = 8.4 Hz, J12b,11 = 5.1 Hz, 1H, H-12b), 3.78–3.85 (m, 2H, H-1a, H-3), 3.76 (dd, J10,11 = 7.3 Hz, J10,9 = 3.7 Hz, 1H, H-10), 3.68 (dd, J1b,1a = 10.5 Hz, J1b,2 = 10.5 Hz, 1H, H-1b), 3.52 (ddd, J = 9.7 Hz, J4,OH = 5.1 Hz, J = 4.4 Hz, 1H, H-4), 3.40 (ddd, J2,1b = 9.9 Hz, J = 9.9 Hz, J = 5.8 Hz, 1H, H-2), 1.45, 1.39, 1.32, 1.31, 1.28, 1.26 (6s, 18H, C(CH3)2).13C-NMR (101 MHz, DMSO-d6): δ(ppm) = 112.5 (C-7), 112.3, 108.2, 98.8 (C(CH3)2), 97.4 (C-6), 83.9 (C-5), 78.2 (C-9), 77.0 (C-8), 76.9 (C-10), 72.7 (C-11), 71.1 (C-3), 67.8 (C-4), 66.8 (C-2), 66.1 (C-12), 61.1 (C-1), 28.9,26.6, 25.8, 25.5, 25.2, 19.0 (C(CH3)2). HRESIMS m/z 483.18377 (calcd for C21H32O11Na, 483.18368); anal. C 54.58, H 7.41, calcd for C21H32O11, C 54.78, H 7.00.



{kind=link}
{kind=link}
{kind=link}

