Planar-Chiral P and N-bridged Diferrocenes



Abstract
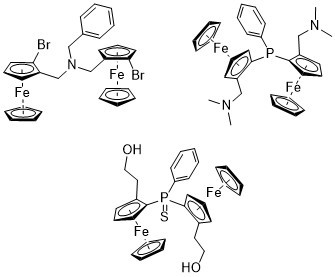
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
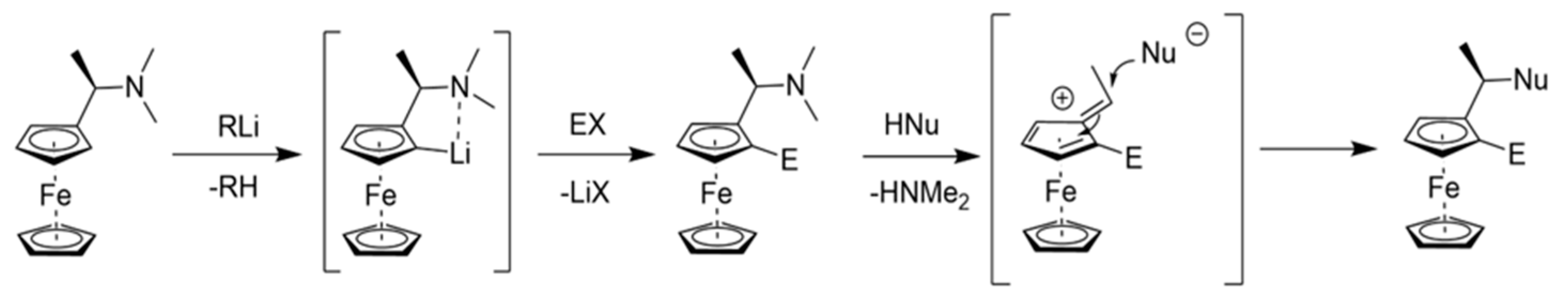
1. Introduction
2. Results
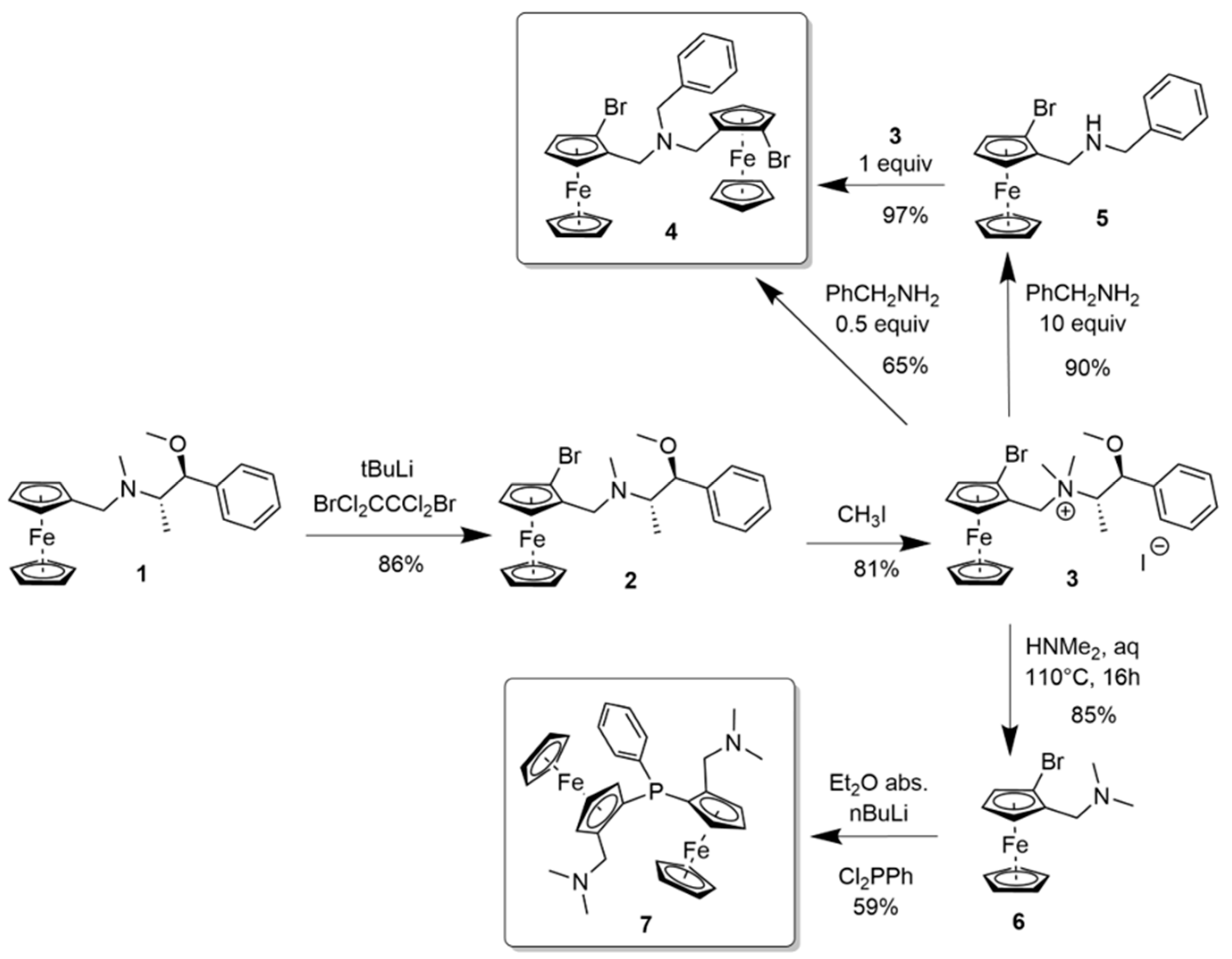
2.1. N-bridged Diferrocenyl Compound 4
2.2. P-bridged Diferrocenyl Compound 7
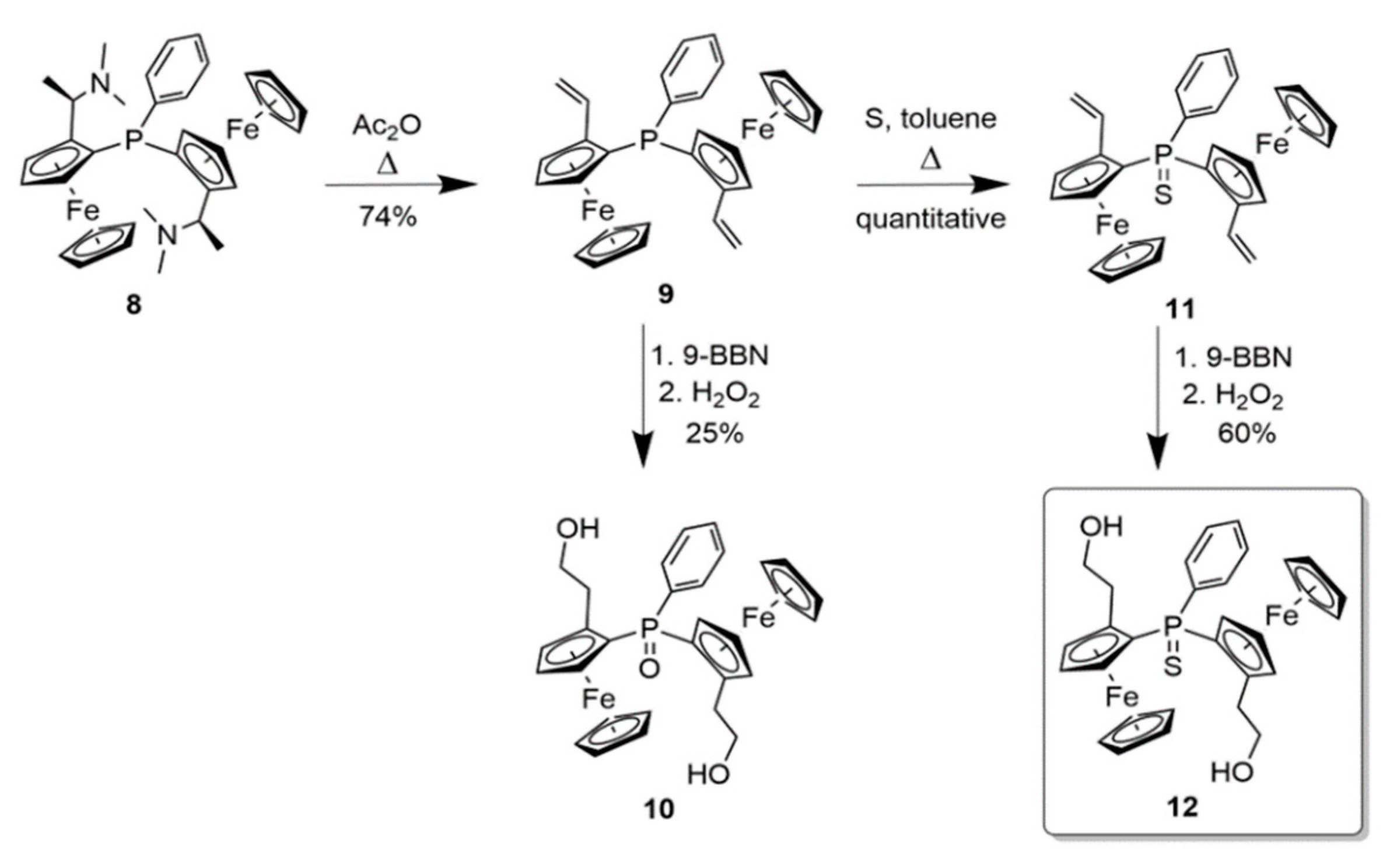
2.3. 1,2-Substituted Diferrocenyl Phosphinsulfide 12
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. N-(2Rp-Bromoferrocenylmethyl)-N-methyl-1-methoxy-1-phenylprop-2-ylamine 2
3.2.2. N,N-Bis(2Rp-bromoferrocenylmethyl)benzylamine 4
3.2.3. N-(2Rp-Bromoferrocenylmethyl)benzylamine 5
3.2.4. 1,1ʺ-(Phenylphosphinidene)bis[(2Rp)-2-[(dimethylamino)-methyl]]ferrocene 7
3.2.5. 1,1ʺ-(Phenylphosphinideneoxide)di[(2Sp)-2-(-1-hydroxy-2-ethyl)]ferrocene 10
3.2.6. 1,1‘‘-(Phenylphosphinidenesulfide)di[(2Sp)-2-vinyl]ferrocene 11
3.2.7. 1,1‘‘-(Phenylphosphinidenesulfide)di[(2Sp)-2-(-1-hydroxy-2-ethyl)]ferrocene 12
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaarschmidt, D.; Lang, H. Selective Syntheses of Planar-Chiral Ferrocenes. Organometallics 2013, 32, 5668–5704. [Google Scholar] [CrossRef]
- Toma, Š.; Csizmadiova, J.; Meciarova, M.; Šebesta, R. Ferrocene phosphane-heteroatom/carbon bidentate ligands in asymmetric catalysis. Dalton Trans. 2014, 43, 16557–16579. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.-X.; Hou, X.-L. Chiral Ferrocenes. In Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Štěpnička, P. Ferrocenes: Ligands, Materials and Biomolecules; John Wiley & Son: Chichester, UK, 2008. [Google Scholar]
- Manoury, E.; Poli, R. Phosphine-Containing Planar Chiral Ferrocenes: Synthesis, Coordination Chemistry and Applications to Asymmetric Catalysis. In Phosphorus Compounds. Catalysis by Metal Complexes; Peruzzini, M., Gonsalvi, L., Eds.; Springer: Dordrecht, the Netherlands, 2011. [Google Scholar]
- Dwadnia, N.; Roger, J.; Pirio, N.; Cattey, H.; Hierso, J.-C. Input of P,N-(phosphanyl, amino)-ferrocene hybrid derivatives in late transition metals catalysis. Coord. Chem. Rev. 2018, 355, 74–100. [Google Scholar] [CrossRef]
- Butt, N.A.; Liu, D.; Zhang, W. The Design and Synthesis of Planar Chiral Ligands and Their Application to Asymmetric Catalysis. Synlett 2014, 25, 615–630. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Pugin, B.; Spindler, F.; Thommen, M. From a chiral switch to a ligand portfolio for asymmetric catalysis. Acc. Chem. Res. 2007, 40, 1240–1250. [Google Scholar] [CrossRef]
- Marquarding, D.; Klusacek, H.; Gokel, G.; Hoffmann, P.; Ugi, I. Stereoselective syntheses. VI. Correlation of central and planar chirality in ferrocene derivatives. J. Am. Chem. Soc. 1970, 92, 5389–5393. [Google Scholar] [CrossRef]
- Gokel, G.; Marquarding, D.; Ugi, I. Stereoselective syntheses. VIII. Retentive nucleophilic displacements of.alpha.-substituted alkylferrocenes. J. Org. Chem. 1972, 20, 3052–3058. [Google Scholar] [CrossRef]
- Barbaro, P.; Togni, A. A New Chiral Tridentate Ferrocenyl Ligand. Synthesis and Characterization of Its Palladium(II) and Nickel(II) Complexes. Organometallics 1995, 14, 3570–3573. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Oberhauser, W.; Togni, A. Synthesis and characterization of chiral bis-ferrocenyl triphosphine Ni (II) and Rh (III) complexes and their use as catalyst precursors for acetalization reactions. J. Mol. Catal. 1999, 145, 139–146. [Google Scholar] [CrossRef]
- Fadini, L.; Togni, A. Asymmetric hydroamination of acrylonitrile derivatives catalyzed by Ni(II)-complexes. Tetrahedron: Asymmetry 2008, 19, 2555–2562. [Google Scholar] [CrossRef]
- Fadini, L.; Togni, A. Asymmetric Catalytic Hydroamination of Activated Olefins in Ionic Liquids. Helv. Chim. Acta 2007, 90, 411–424. [Google Scholar] [CrossRef]
- Gischig, S.; Togni, A. Synthesis and Coordination Chemistry of a New Chiral Tridentate PCP N-Heterocyclic Carbene Ligand Based on a Ferrocene Backbone. Organometallics 2004, 23, 2479–2487. [Google Scholar] [CrossRef]
- Gischig, S.; Togni, A. PdII Complexes of Tridentate PCP N-Heterocyclic Carbene Ligands: Structural Aspects and Application in Asymmetric Hydroamination of Cyano Olefins. Eur. J. Inorg. Chem. 2005, 2005, 4745–4754. [Google Scholar] [CrossRef]
- Sadow, A.D.; Togni, A. Enantioselective Addition of Secondary Phosphines to Methacrylonitrile: Catalysis and Mechanism. J. Am. Chem. Soc. 2005, 127, 17012–17024. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, P.; Bianchini, C.; Togni, A. Synthesis and Characterization of Ruthenium (II) Complexes Containing Chiral Bis (ferrocenyl)−P3 or−P2S Ligands. Asymmetric Transfer Hydrogenation of Acetophenone. Organometallics 1997, 16, 3004–3014. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Giambastiani, G.; Togni, A. Ruthenium(II) Complexes with Triphosphane Ligands Combining Planar, Phosphorus, and Carbon Chirality: Application to Asymmetric Reduction of Trifluoroacetophenone. Eur. J. Inorg. Chem. 2003, 2003, 4166–4172. [Google Scholar] [CrossRef]
- Milosevic, S.; Togni, A. Enantioselective 1,3-Dipolar Cycloaddition of C,N-Cyclic Azomethine Imines to Unsaturated Nitriles Catalyzed by NiIIPigiphos. J. Org. Chem. 2013, 78, 9638–9646. [Google Scholar] [CrossRef]
- Lee, H.M.; Bianchini, C.; Jia, G.; Barbaro, P. Styrene Cyclopropanation and Ethyl Diazoacetate Dimerization Catalyzed by Ruthenium Complexes Containing Chiral Tridentate Phosphine Ligands. Organometallics 1999, 18, 1961–1966. [Google Scholar] [CrossRef]
- Walz, I.; Togni, A. Ni(II)-catalyzed enantioselective Nazarov cyclizations. Chem. Comm. 2008, 4315–4317. [Google Scholar] [CrossRef]
- Broggini, D. Phosphine and Carbene Ligands for Asymmetric Catalysis Containing Two Planar-Chiral Ferrocenyl Units. Ph.D. Thesis, Swiss Federal Institute Of Technology Zurich, Zurich, Switzerland, 2003. [Google Scholar]
- Barreiro, E.; Broggini, D.; Adrio, L.; White, A.; Schwenk, R.; Togni, A.; Hii, K. Gold (I) complexes of conformationally constricted chiral ferrocenyl phosphines. Organometallics 2012, 31, 3745–3754. [Google Scholar] [CrossRef]
- Smith, A.M.R.; Rzepa, H.S.; White, A.J.P.; Billen, D.; Hii, K.K. Delineating Origins of Stereocontrol in Asymmetric Pd-Catalyzed α-Hydroxylation of 1,3-Ketoesters. J. Org. Chem. 2010, 75, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Zirakzadeh, A.; Kirchner, K.; Roller, A.; Stöger, B.; Widhalm, M.; Morris, R.H. Iron(II) Complexes Containing Chiral Unsymmetrical PNP′ Pincer Ligands: Synthesis and Application in Asymmetric Hydrogenations. Organometallics 2016, 35, 3781–3787. [Google Scholar] [CrossRef]
- Zirakzadeh, A.; Kirchner, K.; Roller, A.; Stöger, B.; Carvalho, M.; Ferreira, L. Synthesis, coordination behavior and structural features of chiral iron(ii) PNP diferrocene complexes. RSC Adv. 2016, 6, 11840–11847. [Google Scholar] [CrossRef]
- Zirakzadeh, A.; de Aguilar, S.R.M.M.; Widhalm, M.; Mereiter, K.; Kirchner, K. Iron(II) complexes featuring chiral PNNP diferrocene: Synthesis and characterization of potential hydrogenation catalysts. J. Organomet. Chem. 2016, 819, 260–265. [Google Scholar] [CrossRef]
- Honegger, P.; Widhalm, M. (2S,4R,6R,8S)-4,6-Dimethyl-1-phenyl-diferroceno-1-phosphines. Molbank 2019, 4, 1098. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Kitzler, R.; Weissensteiner, W. O-Methylephedrine: A Versatile and Highly Efficient ortho-Directing Group. Synthesis of Enantiopure 1,2-Disubstituted Ferrocene Derivatives. J. Org. Chem. 2001, 66, 8912–8919. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Mise, T.; Fukushima, M.; Kagotani, M.; Nagashima, N.; Hamada, Y.; Matsumoto, A.; Kawakami, S.; Konishi, M.; Yamamoto, K.; et al. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine–Transition Metal Complexes. I. Preparation of Chiral Ferrocenylphosphines. Bull. Chem. Soc. Jpn. 1980, 53, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Butler, I.; Cullen, W.; Rettig, S. Synthesis of derivatives of [.alpha.(dimethylamino)ethyl]ferrocene via lithiation reactions and the structure of 2-[.alpha.-(dimethylamino)ethyl]-1,1’,3-tris(trimethylsilyl)ferrocene. Organometallics 1986, 5, 1320–1328. [Google Scholar] [CrossRef]
- Fadini, L.; Togni, A. Ni (II)-PPP complexes: From the hydroamination of activated olefins to the synthesis of β-amino acids. Chimia 2004, 58, 208–211. [Google Scholar] [CrossRef]
- Honegger, P.; Widhalm, M. (2S,4R,8S)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide. Molbank 2020, 2020, 1105. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; York, E.A.; Liu, S.; Edgar, K.J. Hydroboration–oxidation: A chemoselective route to cellulose ω-hydroxyalkanoate esters. J. Carb. Pol. 2015, 133, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Honegger, P.; Widhalm, M. Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal. Molbank 2019, 4, 1090. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honegger, P.; Scheibelberger, L.; Widhalm, M. Planar-Chiral P and N-bridged Diferrocenes. Molbank 2020, 2020, M1130. https://doi.org/10.3390/M1130
Honegger P, Scheibelberger L, Widhalm M. Planar-Chiral P and N-bridged Diferrocenes. Molbank. 2020; 2020(2):M1130. https://doi.org/10.3390/M1130
Chicago/Turabian StyleHonegger, Philipp, Lukas Scheibelberger, and Michael Widhalm. 2020. "Planar-Chiral P and N-bridged Diferrocenes" Molbank 2020, no. 2: M1130. https://doi.org/10.3390/M1130
APA StyleHonegger, P., Scheibelberger, L., & Widhalm, M. (2020). Planar-Chiral P and N-bridged Diferrocenes. Molbank, 2020(2), M1130. https://doi.org/10.3390/M1130