2. Results and Discussion
Condensation of ethyl pyruvate and a slight excess of (
S)-lactic acid with acid catalysis in pentane under Dean–Stark conditions of azeotropic water removal [
3] gave complete reaction after 12 h. Washing with water to remove the acids, evaporation of the pentane and chromatographic separation of the residue then afforded three compounds (
Scheme 1). The main components were the desired dioxolanone
1 (23%) and its diastereomer
1′ (7%). While the major product
1 was obtained in pure form, the minor isomer
1′ was always contaminated by varying amounts of
1 so the NMR data of
1′ are taken from the mixture. The stereochemistry at the newly formed C-2 centre is assumed to be (
R) for the major isomer
1 since this places the larger ester group
trans to the 5-methyl and it is also consistent with the stereochemistry in product
2 as determined by X-ray diffraction (see below). The
1H-NMR spectrum showed an interesting pattern for the OCH
2 protons which were non-equivalent and coupled to each other, giving a pattern which could be analysed with the aid of a simulation as an AB pattern of quartets
(Supplementary Material S2).
The third product
2 gave similar NMR signals to
1 and
1′ but with an additional lactic acid-derived –OCH(Me)C=O– group present, and this was confirmed by the HRMS. Interestingly the OCH
2 protons of the ester again formed an AB pattern of quartets with identical coupling constants to those observed for
1. We initially suspected that it might have the eight-membered ring structure
2′ (
Figure 2) which could be formed by direct condensation of the two reacting components, but this was not consistent with the results of 2D NMR experiments (see
Supplementary Material Figures S8 and S9). In particular, HMBC showed that one CHMe proton was interacting with both the ethyl ester carbonyl and one other carbonyl, with this second carbonyl rather than the ethyl ester interacting with the quaternary CMe methyl protons. The isomeric dioxolanone stucture
2, with the extra lactic acid unit effectively inserted into the ester group of
1, was however consistent with all the NMR data and the chemical shifts could be fully assigned as shown in
Figure 2.
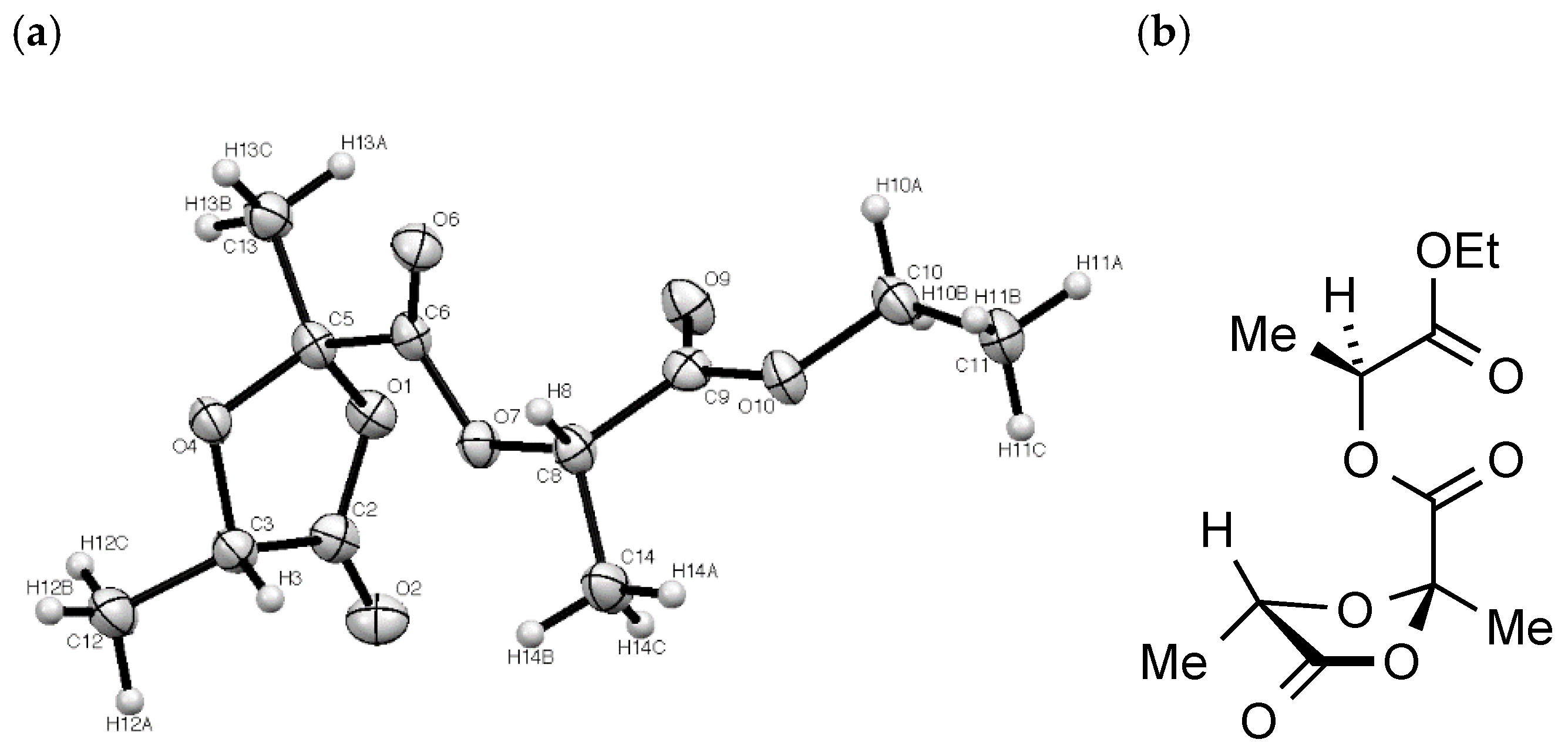
The structure of
2 was confirmed beyond doubt by a single crystal X-ray diffraction study (
Figure 3). In this the dioxolanone ring adopts an approximate envelope conformation with the plane defined by O1-C5-O4 (the flap) at an angle of 22° to the plane of O1-C2-C3-O4. The large ester substituent at C-2 (crystallographic C(5)) then adopts an axial configuration with the methyl equatorial while at C-5 (crystallographic C(3)) the methyl and hydrogen substituents are symmetrically placed above and below the ring.
(a) (b)
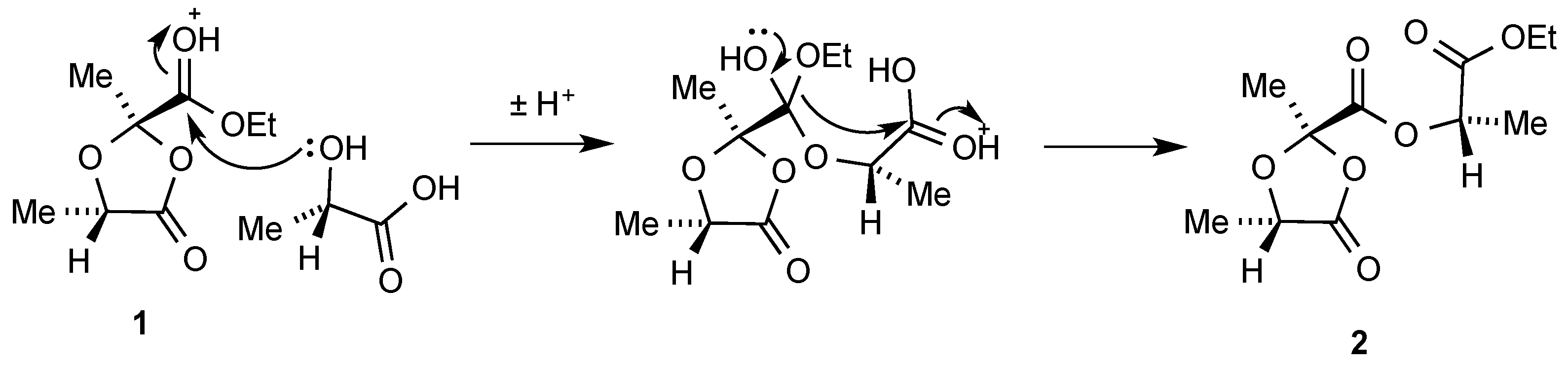
We believe this unexpected product results from reaction of the primary product
1 with a further equivalent of lactic acid. It is tempting to speculate that this may involve an acid-catalysed intramolecular transfer of the ethoxy group as shown in
Scheme 2, although we are unable to exclude the possibility that there is simple transesterification between
1 and lactic acid leading to the dioxolanonecarboxylic acid and ethyl lactate which then undergo condensation.
3. Experimental
Melting points were recorded on a Reichert hot-stage microscope (Reichert, Vienna, Austria) and are uncorrected. IR spectra were recorded on a Perkin-Elmer 1420 instrument (Perkin-Elmer, Waltham, MA, USA). NMR spectra were obtained for protons at 500 MHz and for carbon at 125 MHz using a Bruker AVIII 500 instrument (Bruker, Billerica, MA, USA). Spectra were run at 25 °C on solutions in CDCl
3 with internal Me
4Si as the reference. Chemical shifts are reported in ppm to high frequency of the reference and coupling constants
J are in Hz. Experimental and simulated NMR spectra were processed using i-NMR Reader version 6.1.7 (see
www.inmr.net).
Procedure for synthesis of 1, 1′ and 2: A solution of (S)-lactic acid (90%, 5.0 g, 50 mmol), ethyl pyruvate (5.0 g, 43 mmol), p-toluenesulfonic acid monohydrate (0.05 g, 0.25 mmol) and conc. sulfuric acid (0.25 cm3) in pentane (50 cm3) was heated to reflux with azeotropic removal of water under Dean–Stark conditions for 12 h. The reaction mixture was cooled and washed with water (3 × 20 cm3). The organic layer was separated, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The crude residue was purified by column chromatography (SiO2, 2:1 hexane: diethyl ether as eluent) to give:
Ethyl (2R,5S)-2,5-dimethyl-1,3-dioxolan-4-one-2-carboxylate 1: The title compound was isolated as a clear colourless liquid (1.86 g, 23%); +17.9 (c 1.0 in CH2Cl2); υmax(cm−1) 2990 (C–H), 1817 (C=O), 1753 (C=O), 1304, 1250, 1146 and 941; 1H-NMR (500 MHz, CDCl3) δH: 4.65 (1H, q, J 6.8, 5-CH), 4.280 and 4.264 (2H, AB pattern of q, JAB 11.0, J 7.0, OCH2), 1.79 (3H, s, 2-CH3), 1.50 (3H, d, J 6.8, 5-CH3) and 1.33 (3H, t, J 7.0, CH2CH3); 13C {1H} NMR (125 MHz, CDCl3) δC: 173.0 (C-4), 167.0 (CO2Et), 104.1 (C-2), 71.2 (5-CH), 62.6 (OCH2), 21.9 (2-CH3), 17.1 (5-CH3) and 13.9 (CH2CH3); HRMS (ESI+) C8H12NaO5 [M + Na]+ found 211.0570, requires 211.0582.
Ethyl (2S,5S)-2,5-dimethyl-1,3-dioxolan-4-one-2-carboxylate 1′: The title compound was isolated as a clear colourless liquid (566 mg, 7%) slightly contaminated by 1; 1H-NMR (500 MHz, CDCl3) δH: 4.59 (1H, q, J 6.8, 5-CH), 4.294 and 4.278 (2H, AB pattern of q, JAB 11.0, J 7.0, OCH2), 1.76 (3H, s, 2-CH3), 1.53 (3H, d, J 6.8, 5-CH3) and 1.34 (3H, t, J 7.1, CH2CH3); 13C {1H} NMR (75 MHz, CDCl3) δC: 172.5 (C-4), 167.3 (CO2Et), 104.7 (C-2), 71.0 (5-CH), 62.6 (OCH2), 22.3 (2-CH3), 17.2 (5-CH3) and 13.7 (CH2CH3).
(S)-1-(Ethoxycarbonyl)ethyl(2R,5S)-2,5-dimethyl-1,3-dioxolan-4-one-2-carboxylate 2: The title compound was isolated as a clear colourless liquid (224 mg, 4%) which gradually crystallised with time to a colourless low melting solid, mp 34–36 °C; υmax(cm−1) 2988 (C–H), 1813 (C=O), 1751 (C=O), 1449, 1248, 1132 and 941; 1H-NMR (500 MHz, CDCl3) δH: 5.15 (1H, q, J 7.0, 2’-CH), 4.66 (1H, q, J 7.0, 5-CH), 4.223 and 4.207 (2H, AB pattern of q, JAB 11.0, J 7.0, OCH2), 1.84 (3H, s, 2-CH3), 1.57 (3H, d, J 7.0, 2’-CH3), 1.52 (3H, d, J 7.0, 5-CH3) and 1.28 (3H, t, J 7.0, CH2CH3); 13C-NMR (125 MHz, CDCl3) δC: 172.8 (C-4), 169.4 (C-1’), 166.5 (2-CO), 103.9 (C-2), 71.3 (C-5), 70.3 (C-2’), 61.8 (CH2CH3), 21.9 (2-Me), 16.9 (5-Me), 16.7 (2’-Me) and 14.0 (CH2CH3); HRMS (ESI+) C11H16NaO7 [M + Na]+ found 283.0786, requires 283.0794.
X-ray structure determination: Crystal data for C
11H
16O
7,
M = 260.24 g mol
–1, colourless prism, crystal dimensions 0.12 × 0.03 × 0.02 mm, orthorhombic, space group P2
12
12
1 (No. 19),
a = 4.8832(7),
b = 7.5868(12),
c = 35.478(6) Å,
V = 1314.4(4) Å
3,
Z = 4,
Dcalc = 1.315 g cm
–3,
T = 93 K,
R1 = 0.0360,
Rw2 = 0.0899 for 2149 reflections with
I > 2σ(
I), and 163 variables. Data were collected using graphite monochromated Mo Kα radiation
λ = 0.71075 Å and have been deposited at the Cambridge Crystallographic Data Centre as CCDC 1989784. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via
http://www.ccdc.cam.ac.uk/getstructures. The structure was solved by direct methods and refined by full-matrix least-squares against F
2 (SHELXL Version 2014/7 [
5]). Hydrogen atoms were assigned riding isotropic displacement parameters and constrained to idealised geometries.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




