Abstract
Herein, we described a new protocol for the asymmetric synthesis of apremilast using tert-butanesulfinamide as a chiral auxiliary. This synthetic route consisted of four steps starting from the commercially available 3-hydroxy-4-methoxybenzaldehyde, and apremilast was accordingly obtained in an overall 56% yield and with 95.5% ee.
1. Introduction
Apremilast (Otezla®) is a small molecule inhibitor of phosphodiesterase 4 (PDE4) [1]. It was approved by the FDA for the treatment of patients with moderate to severe plaque psoriasis who may also receive phototherapy or other treatments for psoriasis [2]. Its mechanism of action as a regulator of inflammatory [3,4] signaling renders it as potentially effective for treating various other disease such as ankylosing spondylitis, Behcet’s disease, atopic dermatitis and ulcerative colitis.
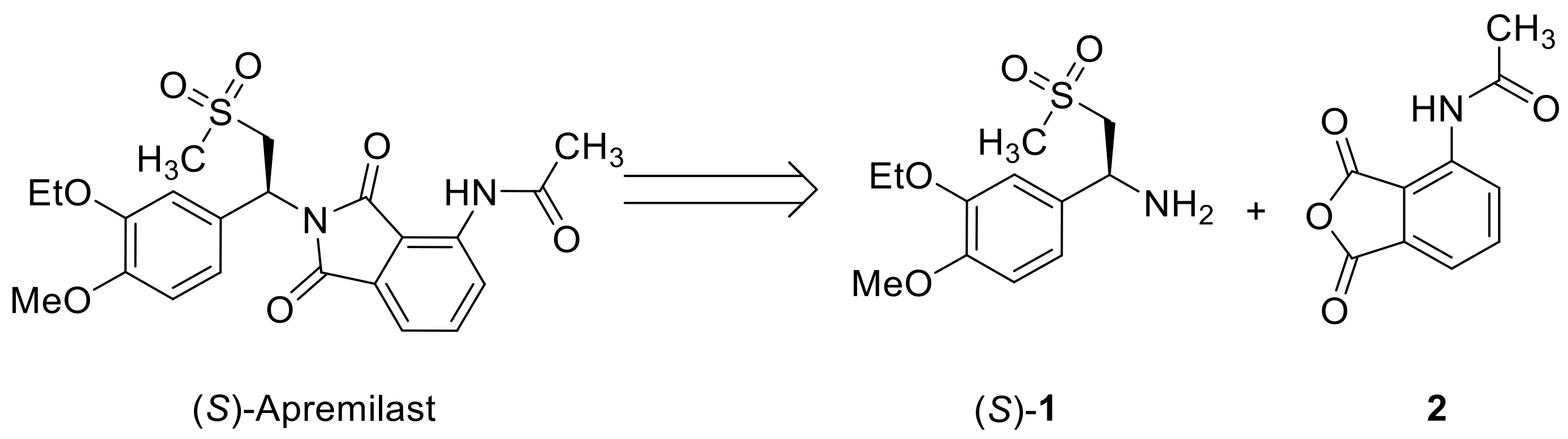
Apremilast possesses the key skeleton of β-amino sulfone (Figure 1), which represents an important class of compounds, interesting for both synthetic chemistry and medicinal applications [5,6]. Notably, the amino group of apremilast in phthalimide form is attached to a benzylic stereogenic carbon. As it has been proven that only the (S)-enantiomer is effective for treating plaque psoriasis [2], it is synthetically demanded to develop efficient methods to prepare (S)-amino sulfone 1, a key intermediate to be coupled with phthalic anhydride 2 for the preparation of apremilast (Figure 1).
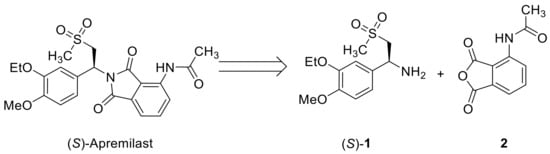
Figure 1.
Structure of apremilast and its key precursors.
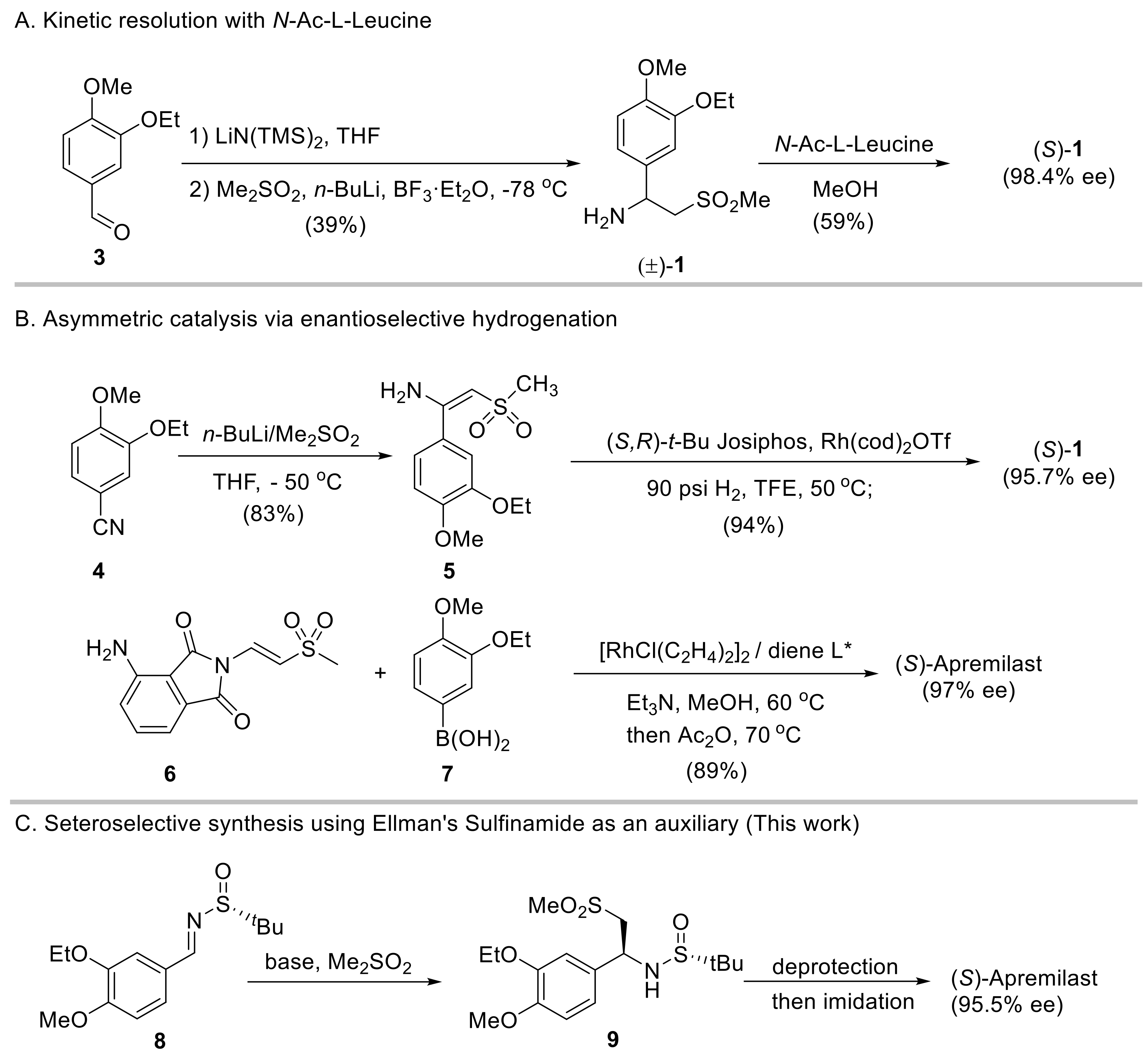
Benzylic amine is common in various pharmaceutical compounds [7,8,9]. The presence of the methylsulfonyl group in the molecule creates some challenges for the asymmetric synthesis of apremilast. For instance, the increasingly popular biocatalytic synthesis by transaminase remains elusive for producing apremilast, despite its wide applications in preparing other chiral aliphatic amines [10,11]. Therefore, the synthesis of apremilast still mainly relies on chemical methods [12]. The discovery approach to access enantioenriched 1 is via kinetic resolution of its racemate mixture by using N-acyl-l-Leucine [13]. Racemate 1 can be obtained by a number of methods, e.g., via base-promoted 1,2-addition of dimethylsulfone (DMS) to the in situ formed imine from aldehyde and LiN(TMS)2 (Scheme 1A). Despite features including a short synthetic sequence and relatively inexpensive reagents, due to the competing double addition of methylsulfone to imines (vide infra) and the waste of the (R)-enantiomer of intermediate 1, this synthetic route is largely concerned with fairly low overall yields.
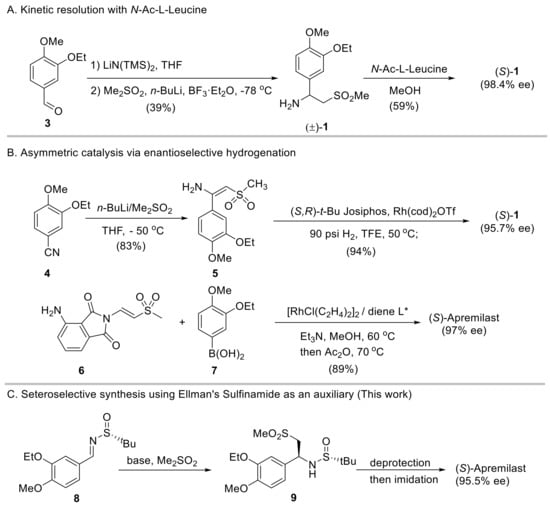
Scheme 1.
Representative synthetic routes to access apremilast.
In recent years, the preparation of chiral amines via asymmetric catalysis has been intensively attempted. In this regard, two representative methods are available for synthesizing the active pharmaceutical ingredient (API) of apremilast. Ruchelman [14] reported a protocol via catalytic asymmetric hydrogenation of sulfonyl enamine 5 with rhodium/(S,R)-tert-Bu Josiphos as the catalyst (Scheme 1B, top). The requisite enamine substrate 5 was prepared through the addition of 3-ethoxy-4-methoxybenzonitrile with the lithium salt of DMS in an 83% yield. The hydrogenation was performed under 90 psi H2 in 2,2,2-trifluoroethanol at 50 °C in the presence of 2 mol% of the chiral rhodium complex, affording chiral amine 1 in a 78% yield (over two steps) and with 95.7% ee. Further upgrading of ee required a resolution process with N-acetyl-l-leucine. A relevant catalytic hydrogenation protocol was later documented by Lv and Zhang using β-acetylamino vinylsulfides as substrates which resulted in chiral β-acetylamino sulfide, a precursor to accessing apremilast involving TaCl5/H2O2 oxidation [15]. More recently, Wu et al. [16] developed a chiral rhodium(I)−diene catalyst enabling the one-step synthesis of β-aryl β-imido sulfones. This complex could be applied to the synthesis of enantioenriched apremilast via the catalytic enantioselective conjugate addition of arylboronic acid 7 to vinyl methyl sulfone 6 in 89% and with 97% ee (Scheme 1B, bottom). Although these approaches have the advantages of high overall yields and excellent asymmetric induction, the expensive starting materials and the latent transition metal residue restrict their industrial application.
Chiral auxiliaries have also been extensively used in the asymmetric synthesis of chiral compounds, including apremilast [12]. Since the advent of enantiopure tert-butanesulfinamide [17] developed by Ellman and co-workers, this sulfur stereogenic center-based chiral auxiliary has proven versatile in diverse asymmetric synthesis [18]. It can undergo efficient condensation with carbonyls under mild conditions and the resulting chiral tert-butanesulfinyl imines then react with different nucleophiles with high stereoselectivity and diastereoselectivity. Herein, we reported our study on the asymmetric synthesis of apremilast via the stereoselective addition of DMS to chiral sulfinyl imine 8 using Ellman’s sulfinamide as an auxiliary (Scheme 1C). Chiral sulfinamide 9 was produced in excellent diastereoselectivity, which led to the presence of apremilast in a high overall yield (56%) and with 95.5% ee.
2. Results and Discussion
2.1. Initial Reaction Attempt and Rational of Outcome
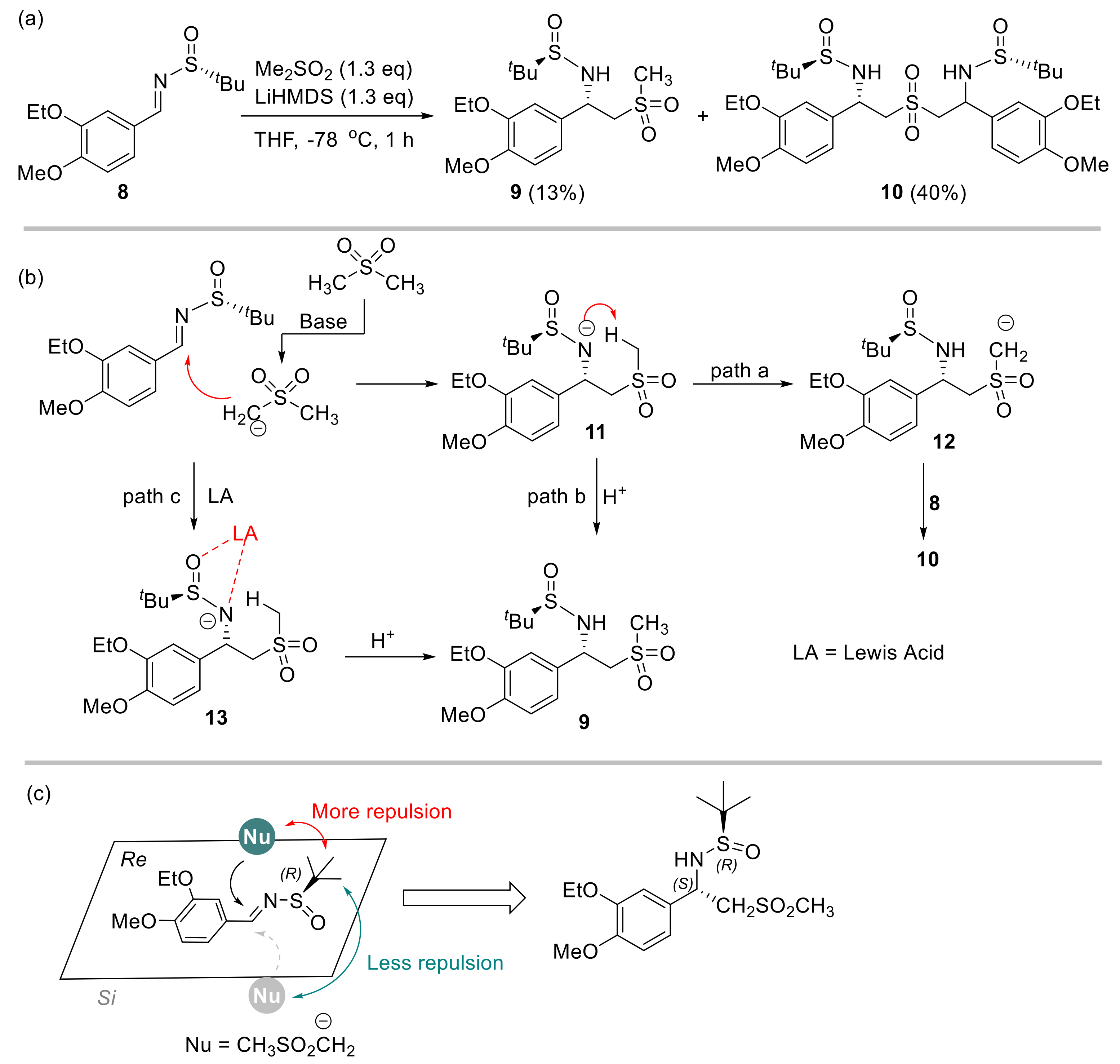
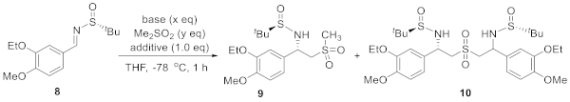
At the outset of our study, (3-ethoxy-4-methoxybenzylidene)-2-methylpropane-2-sulfinamide 8 was treated with the lithium salt of DMS with the intention to obtain chiral sulfinamide 9. This process was inspired by a similar transformation of addition of alkyl phenyl sulfones to N-(tert-butylsulfinyl) aldimines [19]. However, the major product obtained in 40% yield was identified to be the bissulfinamide 10 in addition to the desired adduct 9 (13%, Scheme 2a).
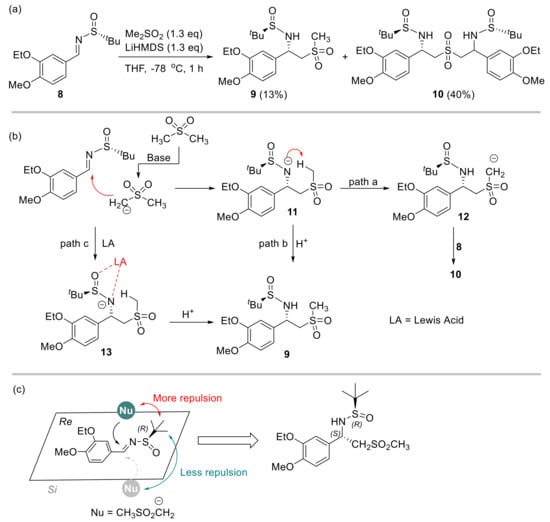
Scheme 2.
Initial reaction outcome and rationale. (a) Addition of DMS to sulfinyl imine 8 promoted by LiHMDS. (b) Rationale of the reaction pathways. (c) A stereochemical model to explain the diastereoselectivity.
Notably, both products were isolated as a single diasteroisomer (d.r. > 25:1) according to NMR analysis of the crude reaction mixture. We speculated that product 10 originated from a competing 1,5-proton transfer of nitrogen anion intermediate 11 to form carbanion 12, followed by the addition of another molecule of sulfinyl aldimine 8 (Scheme 2b, path a). This should be rational because of the deficient external proton source which quenched intermediate 11 and led to the anticipated product 9 (Scheme 2b, path b). To tackle this problem, Lewis acid was employed to stabilize the nitrogen anion to alleviate detrimental intramolecular proton transfer. Meanwhile, increasing the amount of DMS would be beneficial to supply more intermolecular proton source. As such, the formation of product 9 could be significantly enhanced (Scheme 2b, path c) while suppressing the production of side product 10. The excellent diastereoselectivity (d.r. > 25:1) of compound 9 could be explained with the model shown in Scheme 2c. The bulky tert-butyl group provided significant steric repulsion to DMS at the Re face, so this nucleophile had to approach the imine moiety from the Si face. As such, the 1,2-addition proceeded to provide compound 9 with the observed R, S configuration.
2.2. Reaction Condition Optmizations
Keeping the above rationale in mind, we moved on to optimize the reaction conditions by evaluating different bases, additives and their loadings, as well as the amount of DMS (Table 1). The reaction commenced with 1.3 eq. of base, 1.3 eq of DMS in THF at −78 °C. First, it was found that Lithium bis(trimethylsilyl)amide (LiHMDS), sodium bis(trimethylsilyl)amide (NaHMDS), and n-BuLi were all effective in promoting the expected addition reaction with full conversion of substrate 8 in 1 h. However, the ratio of compound 9 to 10 was slightly different and the latter prevailed in all cases (Table 1, entries 1–4). In contrast, major starting materials remained when NaH was used instead, even at 0 °C (Entry 5). As LiHMDS gave superior results, it was employed in the further optimization. As expected, increasing the quantity of DMS was highly beneficial, and the ratio of product 9 to 10 was reversed to 1:0.61 when it was taken in 10.0 eq. of DMS in combination with 5.0 eq. of LiHMDS (Entries 5–7). As DMS was a commonly used solvent, we decided to maintain this loading and, meanwhile, attempt to decrease the dosage of LiHMDS by introducing Lewis acid additives. In this vein, the effect of various Lewis acids (1.0 eq) including MgCl2, AlCl3, FeCl2, NiCl2, BF3, and LiCl was examined (Entries 8–14). The addition of these metal salts did not affect the reaction conversion. Among them, LiCl afforded the optimal outcome and the ratio of product 9 to 10 was elevated to 1:0.15 (Entry 14). Finally, the quantity of the additive was optimized. Pleasingly, the same result was obtained when 0.5 eq of LiCl was used (Entry 15), while the increase in its loading deteriorated the yield of the desired product 9 (Entry 16). Unfortunately, the decrease in the amounts of LiHMDS (e.g., to 1.3 eq) at this point was not encouraged, otherwise the formation of side product 10 would become enhanced (Entry 17).

Table 1.
Reaction condition optimization.
2.3. Synthesis of Apremilast
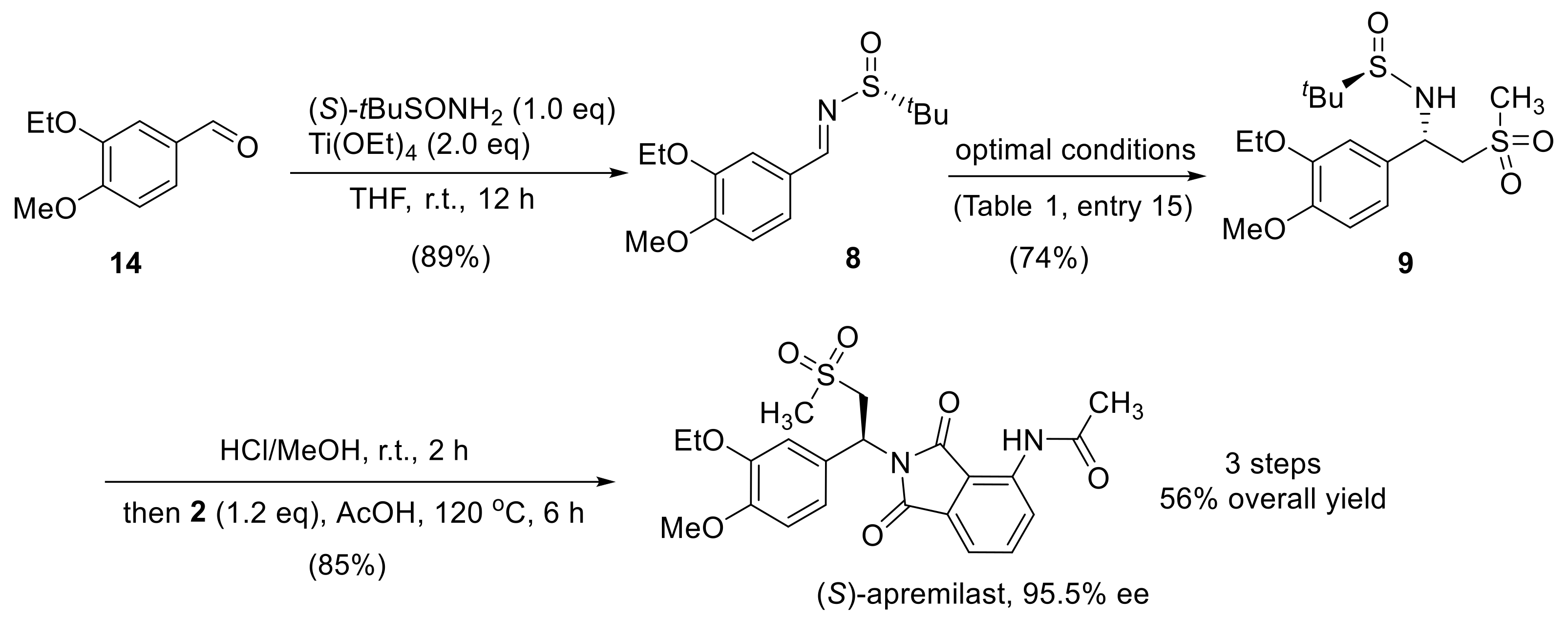
With the optimal reaction condition for the synthesis of compound 9 in hand, the overall synthetic route was established (Scheme 3). Starting from the readily available 3-ethoxy-4-methoxy-benzaldehyde 14, the formation of its tert-butanesulfinyl imine 8 was accomplished with Ti(OEt)4 in an 89% yield. By taking the optimal protocol described above (Table 1, entry 15), intermediate 9 was isolated in a 74% yield with excellent diastereoselectivity (d.r. > 25:1). The removal of the auxiliary was facilely achieved using HCl/MeOH at room temperature to release the free amino group in compound 1, which was readily converted into the apremilast as (S)-enantiomer. The whole synthesis involved four steps with an overall yield of 56%. The final apremilast API was obtained with 95.5% ee.
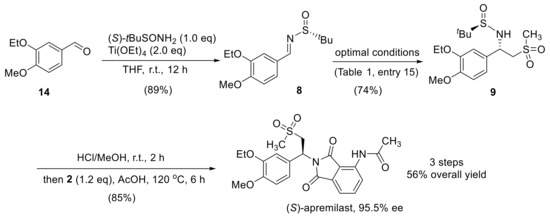
Scheme 3.
Optimized synthetic route for (S)-apremilast.
3. Materials and Methods
3.1. General
Compounds and solvents were purchased from commercial sources and were used as received without further purification unless stated otherwise. All products were purified by flash chromatography on silica gel (200–300 mesh). The chemical yields referred to were isolated products. 1H-NMR and 13C-NMR spectra were recorded on 400 MHz Bruker spectrometers. Chemical shifts were reported in part per million (ppm) relative to residual solvent of CDCl3 (7.26 ppm for 1H-NMR, 77.16 ppm for 13C-NMR). The used abbreviations were as follows: s (singlet), d (doublet), t (triplet), quart. (quartet), quint. (quintet), m (multiplet), br (broad). High resolution mass spectra (HRMS) data were measured on a ESI-microTOF II. Reactions were monitored by TLC analysis using silica gel 60 Å F-254 thin-layer plates and compounds were visualized with a UV light at 254 nm or 365 nm. Further visualization was achieved by staining with iodine, or KMnO4 followed by heating on a hot plate. Flash column chromatography was performed on silica gel 60 Å, 10–40 μm.
3.2. Synthesis of (R,E)-N-(3-Ethoxy-4-Methoxybenzylidene)-2-Methylpropane-2-Sulfinamide (8)
To a flame dried 50 mL flask, (R)-2-methylpropane-2-sulfinamide (1.21 g, 10 mmol, 1.0 eq) and dry THF (15 mL) were added, followed by tetraethyl titanate (4.1 mL, 20 mmol, 2.0 eq). The resulting mixture was stirred for 1 h at room temperature. Then 3-ethoxy-4-methoxybenzaldehyde (1.8 g, 10 mmol, 1.0 eq) dissolved in dry THF (5 mL) was added dropwise into reaction mixture at 0 °C. The reaction mixture was stirred at room temperature for 12 h. After that, 1.0 mL of saturated sodium carbonate solution was added to the mixture. Upon filtration, the residue was then washed with EtOAc. The filtrate was dried over Na2SO4. The crude product was purified by flash column chromatography (Petroleum ether: EtOAc = 20:1) to afford compound 8 (2.7 g, white solid, 90% yield); 1H-NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.39 (dd, J = 8.3, 2.0 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 4.19 (q, J = 7.0 Hz, 3H), 3.96 (s, 3H), 1.52 (t, J = 7.1 Hz, 3H), 1.28 (s, 9H) (Figure S1). 13C-NMR (101 MHz, CDCl3) δ 162.0, 153.1, 148.8, 127.4, 124.8, 111.2, 110.9, 64.4, 57.6, 56.1, 22.6, 14.7 (Figure S2); HRMS (ESI) calcd. For C14H22NO3S [M + H]+: 284.1315, Found 284.1317.
3.3. Synthesis of (R)-N-((S)-1-(3-Ethoxy-4-Methoxyphenyl)-2-(Methylsulfonyl)ethyl)-2-Methylpropane-2-Sulfinamide (9)
To a flame dried 25 mL flask, dimethyl sulfone (0.94 g, 10 mmol, 10 eq), LiCl (22 mg, 0.5 mmol, 0.5 eq) and dry THF (5 mL) were added. The resulting mixture was stirred at −78 °C for 30 min under nitrogen atmosphere. Then LiHMDS (3 mL, 1 M in THF, 3 mmol, 3.0 eq) was added dropwise. Next, the mixture was stirred at −78 °C for another 30 min. Then imine 8 was slowly added (283 mg, 1.0 mmol, dissolved in 4 mL THF) over a period of 10 min and stirred for 1 h at −78 °C. The reaction mixture was quenched by saturated ammonium chloride aqueous and washed with brine. Aqueous phase was extracted three times with EtOAc. The combined organic phases were dried over Na2SO4. The crude product was purified by prepared chromotagraphy (CH2Cl2: CH3OH = 20:1) to afford compound 9 (280 mg, white solid, 74% yield). 1H-NMR (400 MHz, CDCl3) δ 6.97 (m, 2H), 6.90 (d, J = 8.8 Hz, 1H), 4.98 (dt, J = 7.9, 4.8 Hz, 1H), 4.49 (d, J = 4.1 Hz, 1H), 4.14 (q, J = 7.0 Hz, 2H), 3.90 (s, 3H), 3.81 (dd, J = 14.4, 7.5 Hz, 1H), 3.46 (dd, J = 14.3, 5.3 Hz, 1H), 2.71 (s, 3H), 1.49 (t, J = 7.0 Hz, 3H), 1.23 (s, 9H) (Figure S3); 13C-NMR (101 MHz, CDCl3) δ 13C-NMR (101 MHz, CDCl3) δ 149.7, 148.8, 130.7, 119.6, 112.2, 111.6, 64.6, 60.7, 56.4, 56.0, 54.2, 42.5, 22.5, 14.7 (Figure S4). HRMS (ESI) calcd. For C16H28NO5S2 [M + H]+: 378.1403, Found 378.1400.
3.4. Synthesis of (S)-N-(2-(1-(3-Ethoxy-4-Methoxyphenyl) -2-(Methylsulfonyl)ethyl)-1,3-Dioxoisoindolin-4-yl)Acetamide (Apremilast)
To a flask containing (S)-N-((S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl)-2-methylpropane-2-sulfinamide 9 (283 mg, 0.63 mmol), HCl/MeOH solution (12 mL, v/v, 37% HCl/MeOH = 10:1) was added. The resulting reaction mixture was stirred at room temperature for 2 h. MeOH was then removed under vacuum, brine (5 mL) was added to the residue and the mixture was washed three times with ether. The aqueous was basified with sodium hydroxide aqueous (1 M) and then extracted with EtOAc for three times. The combined organic phases were dried over Na2SO4 and concentrated. The crude mixture containing (S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethan-1-amine was dried in a flask, and N-(1,3-dioxo-1,3-dihydroisobenzofuran-4-yl) acetamide (155 mg, 0.76 mmol, 1.2 eq) and glacial acetic acid (4 mL) were added. The reaction mixture was stirred at 120 °C for 6 h. After reaction, the solvent was removed under vacuum. To the residue CH2Cl2 and saturated sodium bicarbonate aqueous were added. The mixture was stirred at room temperature for 15 min and extracted with EtOAc. The combined organic phases were dried over Na2SO4. The crude product was purified by flash chromotagraphy (CH2Cl2) to provide apremilast. (247 mg, yellow solid, 85 % yield). 1H-NMR (400 MHz, CDCl3) δ 9.48 (s, 1H), 8.78 (d, J = 8.4 Hz, 1H), 7.67 (dd, J = 8.5, 7.3 Hz, 1H), 7.51 (dd, J = 7.3, 0.8 Hz, 1H), 7.12 (d, J = 7.3 Hz, 2H), 6.86 (m, 1H), 5.89 (dd, J = 10.5, 4.3 Hz, 1H), 4.58 (dd, J = 14.4, 10.5Hz, 1H), 4.13 (q, J = 7.0 Hz, 2H), 3.87 (s, 3H), 3.74 (dd, J = 14.4, 4.4 Hz, 1H), 2.89 (s, 3H), 2.29 (s, 3H), 1.49 (t, J = 7.0 Hz, 3H) (Figure S5). 13C-NMR (101 MHz, CDCl3) δ 169.6, 169.2, 167.5, 149.8, 148.7, 137.7, 136.2, 131.1, 129.3, 125.0, 120.3, 118.3, 115.2, 112.5, 111.5, 64.6, 56.0, 54.6, 48.6, 41.7, 25.0, 14.7 (Figure S6). The ee was determined on a Chiralpak IA column with hexanes–2-propanol = 8:2, flow = 1.0 mL/min, wavelength = 250 nm. Retention times: 43.7 min (minor), 52.6 min (major). The ee value of synthesized apremilast is 95.5%, which was upgraded to 99.2% after a single recrystallization. Optical rotation: [α]25D = +30.3 (c 1.00, CHCl3). The characterization data are consistent with those reported in the literature [16].
4. Conclusions
In conclusion, we have established a novel synthetic method toward chiral apremilast API compound using Ellman’s Sulfinamide as a chiral auxiliary. The challenge of the process lies in the competing formation of bissulfinamide when performing the ad-dition of DMS to N-(tert-butylsulfinyl) aldimine. This side reaction was effectively suppressed by using excess DMS and the addition of LiCl as Lewis acid to stabilize the sulfinamide anion intermediate. This protocol features the easy availability of starting materials, a short synthetic route, a high overall yield (56% over three synthetic steps starting from commercial available compounds) and an excellent enantioselectivity (95.5% ee) for the product, which holds potential for the industrial preparation of apremilast.
Supplementary Materials
The following are available online, Figure S1: 1H-NMR of (R,E)-N-(3-ethoxy-4-methoxybenzylidene)-2-methylpropane-2-sulfinamide (8); Figure S2: 13C-NMR of (R,E)-N-(3-ethoxy-4-methoxybenzylidene)-2-methylpropane-2-sulfinamide (8); Figure S3: 1H-NMR of (R)-N-((S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl)-2-methylpropane-2-sulfinamide (9); Figure S4: 13C-NMR of (R)-N-((S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl)-2-methylpropane-2-sulfinamide (9); Figure S5: 1H-NMR of Apremilast; Figure S6: 13C-NMR of Apremilast.
Author Contributions
Conceptualization, F.Z.; methodology, B.W.; data curation, B.W.; investigation, B.W.; writing—original draft preparation, B.W.; writing—review and editing, F.Z.; supervision, F.Z.; funding acquisition, F.Z. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Science, Technology and Innovation Commission of Shenzhen Municipality, JCYJ20180305180832515.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to the Analytical and Testing Centre of HUST, Analytical and Testing Centre of School of Chemistry and Chemical Engineering (HUST) for access to their facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Papp, K.; Cather, J.C.; Rosoph, L.; Sofen, H.; Langley, R.G.; Matheson, R.T.; Hu, C.C.; Day, R.M. Efficacy of apremilast in the treatment of moderate to severe psoriasis: A randomised controlled trial. Lancet 2012, 380, 738–746. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA News Release: FDA Approves Otezla to Treat Psoriatic Arthritis. US Department of Health and Human Services. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=205437 (accessed on 25 August 2021).
- Pages, L.; Gavaldà, A.; Lehner, M.D. PDE4 inhibitors: A review of current developments (2005–2009). Expert Opin. Ther. Pat. 2009, 19, 1501–1519. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem. Pharmacol. 2012, 83, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Meadows, D.C.; Gervay-Hague, J. Vinyl sulfones: Synthetic preparations and medicinal chemistry applications. Med. Res. Rev. 2006, 26, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Back, T.G. Design and synthesis of some biologically interesting natural and unnatural products based on organosulfur and selenium chemistry. Can. J. Chem. 2009, 87, 1657–1674. [Google Scholar] [CrossRef]
- Ghislieri, D.; Turner, N.J. Biocatalytic approaches to the synthesis of enantiomerically pure chiral amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Shi, Y.; Zhong, F. Rhodium-catalyzed intermolecular C(sp3)–H amination in a purely aqueous system. Green. Chem. 2018, 20, 113–117. [Google Scholar] [CrossRef]
- Michael, F.; Farnberger, J.E.; Kroutil, W. The Industrial Age of Biocatalytic Transamination. Eur. J. Org. Chem. 2015, 32, 6965–6982. [Google Scholar]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S.; Gilmore, B.F. Application of ω-Transaminases in the Pharmaceutical Industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Narode, H.; Gayke, M.; Eppa, G.; Yadav, J.S. A Review on Synthetic Advances toward the Synthesis of Apremilast, an Anti-inflammatory Drug. Org. Process. Res. Dev. 2021, 25, 1512–1523. [Google Scholar] [CrossRef]
- Man, H.-W.; Schafer, P.; Wong, L.M.; Patterson, R.T.; Corral, L.G.; Raymon, H.; Blease, K.; Leisten, J.; Shirley, M.A.; Tang, Y.; et al. Dis-covery of (S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide (Apremilast), a Potent and Orally Active Phosphodiesterase 4 and Tumor Necrosis Factor-α Inhibitor. J. Med. Chem. 2009, 52, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Ruchelman, A.L.; Connolly, T.J. Enantioselective synthesis of the apremilast aminosulfone using catalytic asymmetric hydrogenation. Tetrahedron Asymmetry 2015, 26, 553–559. [Google Scholar] [CrossRef]
- Gao, W.; Lv, H.; Zhang, X. Rh/DuanPhos-Catalyzed Asymmetric Hydrogenation of β-Acetylamino Vinylsulfides: An Approach to Chiral β-Acetylamino Sulfides. Org. Lett. 2017, 19, 2877–2880. [Google Scholar] [CrossRef] [PubMed]
- Syu, J.-F.; Gopula, B.; Jian, J.-H.; Li, W.-S.; Kuo, T.-S.; Wu, P.-Y.; Henschke, J.P.; Hsieh, M.-C.; Tsai, M.-K.; Wu, H.-L. Asymmetric Synthesis of β-Aryl β-Imido Sulfones Using Rhodium Catalysts with Chiral Diene Ligands: Synthesis of Apremilast. Org. Lett. 2019, 21, 4614–4618. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cogan, D.A.; Ellman, J.A. Catalytic Asymmetric Synthesis of tert-Butanesulfinamide. Application to the Asymmetric Synthesis of Amines. J. Am. Chem. Soc. 1997, 119, 9913–9914. [Google Scholar] [CrossRef]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Xu, W.; Zheng, W.; Zhou, P.; Sun, Z. Practical and stereoselective synthesis of β-amino sulfones from alkyl phenyl sulfones and N-(tert-butylsulfinyl) aldimines. Org. Biomol. Chem. 2011, 9, 6502–6505. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).