2. Results and Discussion
Aromatic nucleophilic substitution of 4,7-dibromo[1,2,5]thiadiazolo[3,4-
d]pyridazine with thiols led to the formation of 4,7-bis-thiosubstituted [1,2,5]thiadiazolo[3,4-
d]pyridazines [
4]; all attempts to isolate mono-substituted derivatives were unsuccessful. On the other hand, for the synthesis of 4,7-bis(alkylthio)benzo[
c][1,2,5]thiadiazole, Buchwald–Hartwig conditions are required: treatment with alkylthiols and a mixture of Pd
2(dba)
3, DPPF, and DIPEA [
2]. We assumed that the synthesis of 4,7-bis(dodecylthio)-[1,2,5]thiadiazolo[3,4-
c]pyridine
1 from 4,7-dibromo-[1,2,5]thiadiazolo[3,4-
c]pyridine
2 can proceed smoothly through a combination of the two reactions—nucleophilic substitution and Buchwald–Hartwig cross-coupling.
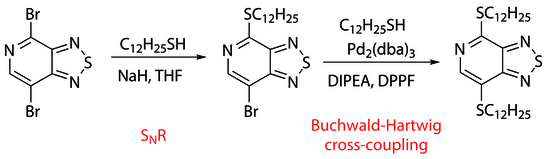
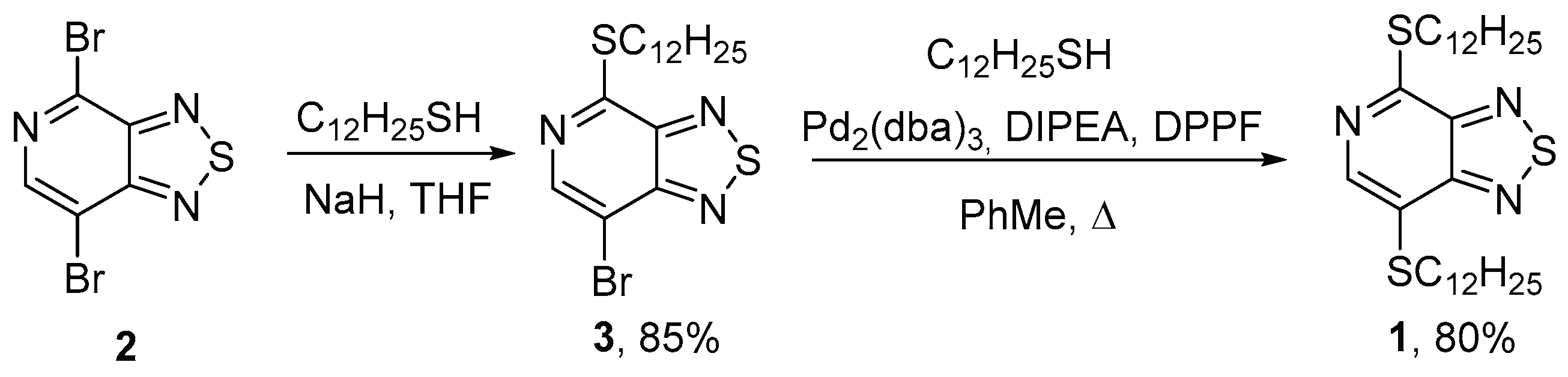
Dibromide
2 was studied in the reaction of aromatic nucleophilic substitution S
NAr with dodecane-1-thiol (
Scheme 1,
Table 1). It was shown that when dibromide
2 was treated with two equivalents of thiol at room temperature in various organic solvents (CHCl
3, THF, MeCN, and DMF), only monomercapto derivative
3 was formed (
Table 1, entries 1–4). The reaction in an aprotic dipolar solvent (DMF) proceeded much faster than in a less polar organic solvent, chloroform. We showed that the use of sodium thiolate led to an increase in the yield of dodecylthio derivative
3 up to 85% (
Table 1, entry 5). Previously, it has been shown that the S
NAr reactions with 4,7-dibromo[1,2,5]thiadiazolo[3,4-
c]pyridine proceed exclusively at the more electron-deficient position 4, which is closest to the pyridine nitrogen atom [
5].
To synthesize the target dithiol 1, we investigated the Buchwald–Hartwig cross-coupling of monothiol 3 with dodecane-1-thiol in the presence of a palladium catalyst tris(dibenzylideneacetone)dipalladium (0) (Pd2(dba)3), a DPPF ligand, and DIPEA as a base. It was found that when the reaction mixture was refluxed in toluene for 6 h, the starting derivative 3 completely disappeared with the formation of 4,7-bis(dodecylthio)-[1,2,5]thiadiazolo[3,4-c]pyridine 1 in a high yield. The structures of 7-bromo-4-(dodecylthio)-[1,2,5]thiadiazolo[3,4-c]pyridine 3 and 4,7-bis(dodecylthio)-[1,2,5]thiadiazolo[3,4-c]pyridine 1 were confirmed by means of elemental analysis; high-resolution mass spectrometry; 1H, 13C NMR, IR and UV spectroscopy; and mass spectrometry.
In conclusion, 4,7-bis(dodecylthio)-[1,2,5]thiadiazolo[3,4-c]pyridine 1 was successfully prepared from 4,7-dibromo-[1,2,5]thiadiazolo[3,4-c]pyridine 2 by combining two synthetic procedures: aromatic nucleophilic substitution SNAr and Buchwald–Hartwig cross-coupling reaction. The liquid crystalline properties of bis(dodecylthio) derivative are being investigated.
3. Materials and Methods
4,7-Dibromo-[1,2,5]thiadiazolo[3,4-
c]pyridine
1 was prepared according to the published method [
6]. The solvents and reagents were purchased from commercial sources and used as received. The melting point was determined on a Kofler hot-stage apparatus and was uncorrected.
1H and
13C NMR spectra were taken with a Bruker AM-300 machine (Bruker AXS Handheld Inc., Kennewick, WA, USA) (at frequencies of 300 and 75 MHz) in CDCl
3 solution, with TMS as the standard. J values are given in Hz. The MS spectrum (EI, 70 eV) was obtained with a Finnigan MAT INCOS 50 instrument (Hazlet, NJ, USA). The IR spectrum was measured with a Bruker “Alpha-T” instrument (Santa Barbara, CA, USA) in KBr pellet. The high-resolution MS spectrum was measured on a Bruker micrOTOF II instrument (Bruker Daltonik Gmbh, Bremen, Germany) using electrospray ionization (ESI). Solution UV–visible absorption spectra were recorded using an OKB Spektr SF-2000 UV/Vis/NIR spectrophotometer (St. Petersburg, Russia) controlled with SF-2000 software (St. Petersburg, Russia). The sample was measured in a 1 cm quartz cell at room temperature with 4.8 × 10
−5 mol/mL concentration in CH
2Cl
2.Sodium hydride (23 mg, 1 mmol) was added to a solution of dodecane-1-thiol (202 mg, 1 mmol) in dry THF (30 mL) at 0 °C with stirring. The reaction mixture was stirred at 0 °C for 30 min, then 4,7-dibromo-[1,2,5]thiadiazolo[3,4-c]pyridine 2 (295 mg, 1 mmol) was added. The mixture was stirred for 3 h at room temperature. On completion (monitored by TLC), the mixture was poured into water (20 mL) and extracted with EtOAc (3 × 35 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (Silica gel Merck 60, eluent hexane–CH2Cl2, 5:1, v/v). Yield 353 mg (85%), green solid, mp = 54–56 °C;, Rf = 0.2 (CH2Cl2, 5:1, v/v). IR spectrum, ν, cm–1: 2935, 2844, 1466, 1448, 1392, 1353, 1294, 1254, 1222, 1131, 1104, 1006, 906, 854, 695, 524. 1H NMR (ppm): δ 8.48 (1H, s), 3.37 (t, J = 7.3, 2H), 1.81 (p, J = 7.4, 2H), 1.58–1.45 (m, 2H), 1.39–1.23 (m, 16H), 0.89 (t, J = 5.7, 3H). 13C NMR (ppm): δ 157.3, 154.8, 149.3, 145.4, 106.0, 31.9, 29.8, 29.63, 29.62, 29.58, 29.48, 29.34, 29.16, 28.9, 28.8, 22.6, 14.1. HRMS (ESI-TOF), m/z: calcd. for C17H2779BrN3S2 [M + H]+ 416.0824, found, 416.0809. MS (EI, 70eV), m/z (I, %): 419 ([M + 3]+, 9), 418 ([M + 2]+, 37), 417 ([M + 1]+, 30), 416 ([M]+, 38), 415 ([M − 1]+, 27), 247 (70), 41 (100).
7-Bromo-4-(dodecylthio)-[1,2,5]thiadiazolo[3,4-c]pyridine 2 (300 mg, 0.72 mmol), Pd2(dba)3 (6 mg, 1 mmol%), DPPF (8 mg, 2 mmol%) and DIPEA (0.13 mL, 0.79 mmol) were dissolved in a vial with 10 mL toluene under a stream of nitrogen. After 10 min, dodecane-1-thiol (145 mg, 0.17 mL, 0.72 mmol) was added using a syringe. The temperature of the oil bath was increased to 120 °C, and stirring was continued for 6 h. The reaction mixture was poured into ice-water and extracted by CHCl3 (3 × 10 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (Silica gel Merck 60, eluent hexane–CH2Cl2, 5:1, v/v). Yield 353 mg (85%), yellow solid, mp = 89–91 °C;, Rf = 0.3 (CH2Cl2, 5:1, v/v). IR spectrum, ν, cm–1: 2956, 2919, 2849, 1540, 1467, 1442, 1299, 1250, 1242,991, 956, 879, 870, 721, 631, 562, 493. 1H NMR (ppm): δ 8.28 (1H, s), 3.37 (t, J = 7.3, 2H), 3.11 (t, J = 7.4, 2H), 1.87–1.77 (m, 2H), 1.72–1.63 (m, 2H), 1.54–1.42 (m, 4H), 1.33–1.23 (m, 32H), 0.89 (t, J = 6.5, 6H). 13C NMR (ppm): δ 156.3, 155.8, 149.3, 144.1, 120.1, 33.2, 2 × 32.0, 29.74, 29.72, 2 × 29.71, 29.70, 29.69, 29.65, 29.60, 29.5, 2 × 29.4, 29.3, 29.28, 29.2, 29.1, 29.0, 28.8, 2 × 22.3, 2 × 14.2. HRMS (ESI-TOF), m/z: calcd. for C29H52N3S3 [M + H]+ 538.3318, found, 538.3308. MS (EI, 70 eV), m/z (I, %): 537 ([M]+, 83), 490 (20), 369 (30), 43 (100). UV–Vis spectra (in CH2Cl2), λmax: 258 nm (ε = 24705 M−1 cm−1), 427 nm (ε = 7294 M−1 cm−1).







{kind=link}
{kind=link}