3. Materials and Methods
General Experimental Conditions and Materials: Bulk solvents were purchased from Fisher Chemical and Macron; general chemicals were purchased from Acros Organics, Alfa Aesar, Fisher Chemical, Macron, Mallinckrodt Specialty Chemicals, Oakwood Chemical, Sigma-Aldrich, and TCI America and were used as received. SiliaFlash P60 silica gel was purchased from Silicycle. Nitromethane was purchased from Alfa Aesar. 4-Bromoanthranilic acid was purchased from Oakwood Chemical. Sodium dithionite was purchased from J.T. Baker. Valeryl chloride and 9,9-dimethyl-9
H-xanthene-4,5-diylbis(diphenylphosphane) (XantPhos) were purchased from TCI America. Benzoyl isocyanate and phenol were purchased from Sigma-Aldrich. Tris(dibenzylideneacetone)dipalladium was purchased from Chem-Impex. An anhydrous solvent dispensing system (MBraun MB-SPS-800 or Inert PureSolv MD 5) was used for drying toluene and this solvent was dispensed under nitrogen. Thin-layer chromatography (TLC) was performed on 0.25 mm hard-layer silica GF plates. Developed plates were visualized with a hand-held UV lamp. Column chromatography conditions were based on TLC results. Some
1H NMR spectra were recorded on a Varian 400 MHz spectrometer with a dual broadband probe, while some
1H NMR and
13C{
1H} NMR spectra were recorded on a Bruker 600 MHz Avance NEO spectrometer with a 5 mm triple resonance cryoprobe. Proton chemical shifts are reported in ppm from internal standards of residual chloroform (CDCl
3-7.26 ppm) or dimethylsulfoxide ((CD
3)
2SO-2.50 ppm).
13C{
1H} chemical shifts are reported in ppm from internal standards of residual chloroform (CDCl
3-77.0 ppm) or dimethylsulfoxide ((CD
3)
2SO-39.5 ppm). Proton chemical data are reported as follows: chemical shift (multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet, br = broad, prefixed “app-” = apparent), coupling constant, and integration). High-resolution mass spectra were obtained on an Agilent TOF II TOF/MS instrument, equipped with an ESI or APCI interface or on a Bruker BioTOF II with an ESI interface. (See
Supplementary Materials for
1H NMR,
13C{
1H} NMR, and HRMS spectra.)
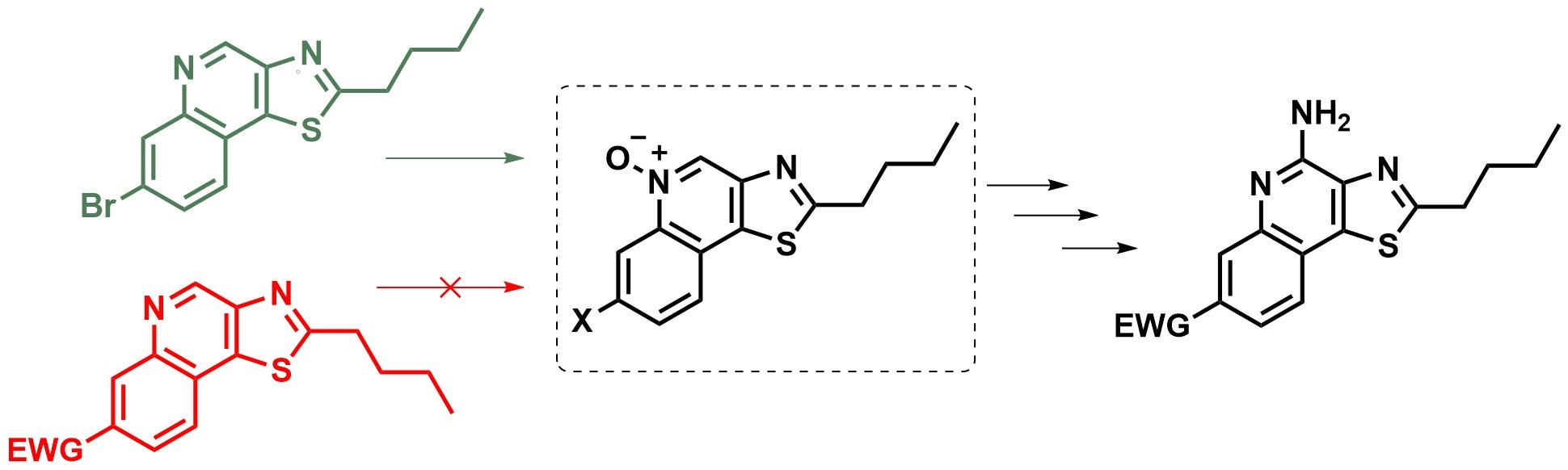
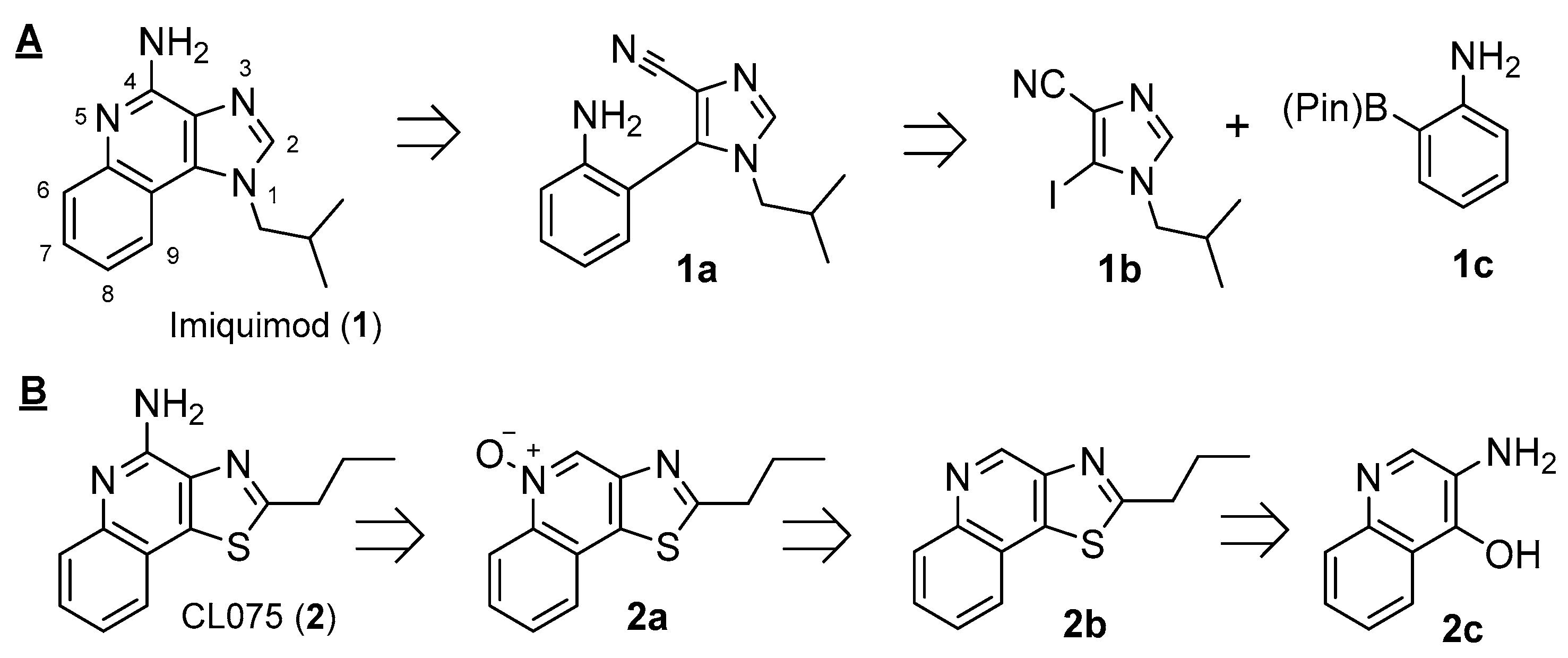
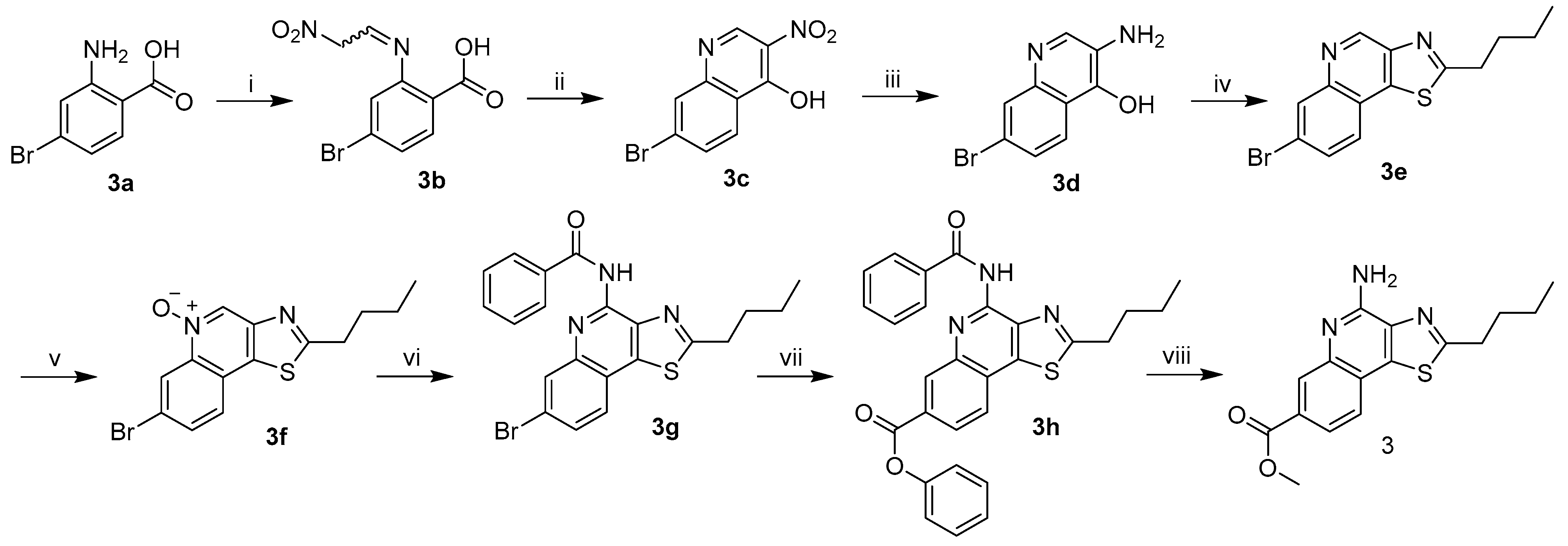
The preparation of methazoic acid was adapted from established methods [
10]. Sodium hydroxide (6.72 g, 168 mmol) was dissolved in water (12 mL) and maintained at 25 °C. To this aqueous sodium hydroxide, nitromethane (3 mL, 56 mmol) was added dropwise with rapid stirring and heated to 35 °C for 10 min. The reaction was cooled to 25 °C and another portion of nitromethane (3 mL, 56 mmol) was added while carefully maintaining the temperature at or below 35 °C. After 10 min of rapid stirring, the reaction was heated to 50 °C for 5 min and then allowed to cool to 25 °C over 10 min. The reaction mixture was poured into cold water (50 mL) and concentrated hydrochloric acid (14 mL, 167 mmol) was then added. Additional water was added until the total volume reached 80 mL. The crude methazoic acid solution should be used immediately in the following step or frozen (−20 °C) and carefully thawed just before use. (Note: Although dilute methazoic acid or sodium methazoate is typically not a shock or temperature sensitive explosive, methazoic acid containing salts or solutions can be explosive).
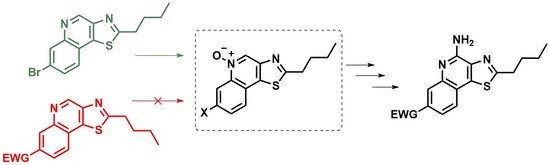
4-Bromo-2-((2-nitroethylidene)amino)benzoic acid (3b). To a fully dissolved solution of 4-bromoanthranilic acid (3.0 g, 13.89 mmol, 1 equiv) in a 1:1 mixture of water:dioxane (90 mL) and concentrated hydrochloric acid (1.2 mL, 14 mmol, ~1 equiv) at 50 °C, was added the crude aqueous methazoic acid solution (40 mL, ~28 mmol, ~2 equiv), freshly prepared as described above. An off-white precipitate immediately began to form and the reaction was allowed to cool to 25 °C and stirred for 18 h. The pale-yellow precipitate was filtered and rinsed with water until the filtrate was clear. The filtered solid was dried to constant mass (3.1 g, 78%) in a vacuum oven (50 °C, ~30 mbar). This can be recrystallized from 45 mL of 1:2 EtOAc:MeOH / gram of crude product to reduce the presence of impurities in later intermediates, but recrystallization did not have a consistent effect on the yield of the next reaction. The recrystallized solid has a stronger yellow color. The purity was qualitatively shown to have improved by TLC. The solid was carried forward without additional characterization: Rf = 0.15 (97:2:1 EtOAc:MeOH:H2O).
7-Bromo-3-nitroquinolin-4-ol (3c). 3b (0.65 g, 2.28 mmol, 1 equiv) was added to a flask with stir bar and acetic anhydride (7.5 mL). This suspension was heated to 100 °C for 45 min or until fully dissolved. Anhydrous sodium acetate (0.51 g, 6.27 mmol, 2.75 equiv) was added to the reaction in one portion while vigorously stirring. The reaction was heated to reflux for 1 h and then allowed to cool to 25 °C. The contents of the flask were diluted with cooled 80% aqueous acetic acid and the solid was filtered. The solid was rinsed with acetic acid until the filtrate was clear. The solid was dried in a vacuum oven (50 °C, ~30 mbar) for 18 h to give a tan powder (0.35 g, 57%) that easily lifted off of filter paper. The crude solid exhibited poor solubility in most solvents and was carried on without characterization.
3-Amino-7-bromoquinolin-4-ol (3d). To a solution of crude 3c (0.22 g, 0.82 mmol, 1 equiv) in anhydrous DMF (15 mL) was added sodium dithionite (1.15 g, 6.6 mmol, 8 equiv). The reaction mixture was stirred for 18 h under N2 at 25 °C. DMF was removed in vacuo and the crude solid was suspended in ethyl acetate (20 mL) and then washed with hot water (20 mL). The organic layer was clear and the aqueous layer was dark brown. This mixture was stirred and heated to 70 °C while solid sodium carbonate powder (2.5 g) was added and allowed to fully dissolve. The organics (now brown) and solids at the interface were separated from the aqueous layer (a lighter yellowish brown). The aqueous layer was carefully extracted with ethyl acetate (4 × 10 mL). The product exhibited reasonable solubility in the aqueous layer. Organics (with solids) were combined and concentrated in vacuo. The crude compound was purified by silica column 85:9:3:3 EtOAc:MeOH:H2O:Et3N to yield 0.09 g (46%) of 3d as an off-white solid: 1H NMR ((CD3)2SO, 400 MHz) δ 11.46 (br s, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.67 (s, 1H), 7.53 (s, 1H), 7.28 (d, J = 8.8 Hz, 1H), 4.53 (br s, 2H). 13C{1H} NMR ((CD3)2SO, 600 MHz) δ 169.2, 138.0, 131.9, 127.1, 124.1, 122.8, 120.3, 120.0, 119.3. (ESI+): Calcd C9H8BrN2O [M + H] + 238.9815, found 238.9810 (error 1.8 ppm).
7-Bromo-2-butylthiazolo[4,5-c]quinoline (3e). To a solution of 3d (0.07 g, 0.29 mmol, 1 equiv) in anhydrous pyridine (3.4 mL), being purged with N2, was added valeryl chloride (0.044 g, 0.37 mmol, 1.25 equiv). This mixture was heated to 60 °C for 2 h in a pressure vessel. The reaction was cooled to 25 °C and again placed under a stream of N2. Phosphorus pentasulfide (0.14 g, 0.32 mmol, 1.1 equiv) was added and the reaction was heated to 105 °C for 18 h in the pressure vessel. The reaction was cooled to 25 °C and aqueous sodium carbonate (10 mL) and ethyl acetate (10 mL) were added. The mixture was separated and the aqueous layer extracted with ethyl acetate (2 × 5 mL). (Caution: there will be trace hydrogen sulfide in the organics. Pulling a light vacuum with some gentle heating for 2 h will usually remove it). The organics were combined and concentrated in vacuo and purified by silica column 1:99 MeOH:DCM to give 0.08 g (86%) of 3e as a yellow waxy solid: 1H NMR (CDCl3, 400 MHz) δ 9.43 (s, 1H), 8.43 (d, J = 1.6 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.73 (dd, J = 8.4, 1.6 Hz, 1H), 3.23 (t, J = 7.6 Hz, 2H), 1.94 (quint, J = 7.6 Hz, 2H), 1.51 (app-sext, J = 7.6, 7.2 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 173.3, 147.9, 146.5, 144.7, 140.5, 132.8, 130.8, 126.0, 122.4, 122.1, 34.0, 31.7, 22.2, 13.7. (ESI+): Calcd C14H14BrN2S [M + H] + 321.0056, found 321.0068 (error 3.8 ppm).
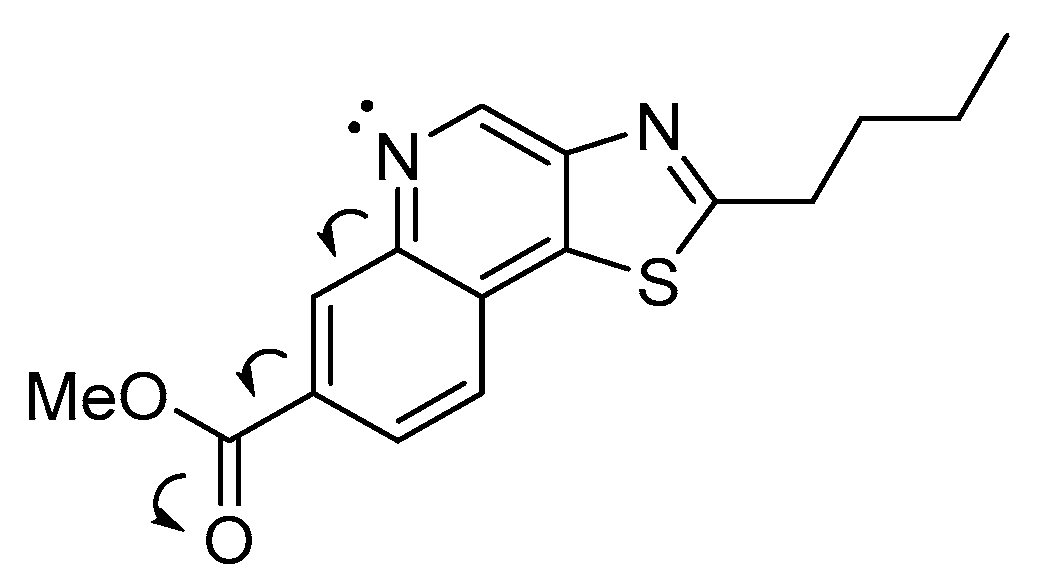
7-Bromo-2-butylthiazolo[4,5-c]quinoline 5-oxide (3f). To a solution of 3e (0.075 g, 0.23 mmol, 1 equiv) in dichloromethane (3 mL) was added 53% m-chloroperoxybenzoic acid (0.27 g, 0.82 mmol, 3.5 equiv). This was stirred at 25 °C for 15 h. TLC indicated a near-complete reaction. The reaction mixture was washed with saturated aqueous sodium carbonate (10 mL) and diluted with an additional portion of dichloromethane (10 mL) and water (10 mL) before the mixture was separated, the aqueous layer was extracted with DCM (2 × 10 mL), and the organics were combined and concentrated in vacuo. The crude solid 3f was purified by silica column 1:99–2:98 MeOH:DCM (linear gradient) to give 0.073 g (93%) of a white powder: 1H NMR (CDCl3, 400 MHz) δ 9.19 (s, 1H), 9.11 (s, 1H), 7.84 (s, 2H), 3.20 (t, J = 7.6 Hz, 2H), 1.92 (quint, J = 7.6 Hz, 2H), 1.51 (sext, J = 7.6 Hz, 2H), 1.00 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 175.6, 147.2, 139.2, 133.3, 132.1, 130.6, 126.7, 124.3, 124.0, 122.5, 34.1, 31.7, 22.2, 13.7. (ESI+): Calcd C14H13BrN2OSNa [M + Na] + 358.9824, found 358.9809 (error 4.4 ppm).
N-(7-Bromo-2-butylthiazolo[4,5-c]quinolin-4-yl)benzamide (3g). To a solution of 3f (0.073 g, 0.22 mmol, 1 equiv) in dichloromethane (8 mL) was added benzoyl isocyanate (0.127 g, 0.86 mmol, 4 equiv). The solution turned a light-yellow color after the benzoyl isocyanate was fully dissolved and was stirred at 25 °C for 18 h. The reaction mixture was concentrated in vacuo and purified by silica column 0:100–1:99 MeOH:DCM (linear gradient) which resulted in 0.070 g (73%) of 3g as a white solid: 1H NMR (CDCl3, 400 MHz) δ 9.69 (br s, 1H), 8.41 (s, 1H), 8.08 (s, 1H), 8.06 (s, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.65–7.53 (m, 4H), 3.21 (t, J = 7.6 Hz, 2H), 1.92 (quint, J = 7.6 Hz, 2H), 1.53 (app-sext, J = 7.6, 7.2 Hz, 2H), 1.02 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 173.0, 164.6, 145.4, 144.3, 140.7, 139.3, 134.7, 132.4, 132.2, 129.2, 128.9, 127.7, 125.5, 122.9, 120.2, 34.0, 31.9, 22.3, 13.8. (ESI+): Calcd C21H19BrN3OS [M + H] + 440.0427, found 440.0437 (error 2.3 ppm).
N-(2-Butyl-7-phenoxycarbonylthiazolo[4,5-c]quinolin-4-yl)benzamide (3h). A solution of formic acid (2 mL) in acetic anhydride (5 mL) was stirred at 35 °C for 1 h to generate acetic formic anhydride. 3g (0.07 g, 0.16 mmol, 1 equiv), phenol (0.061 g, 0.65 mmol, 4.1 equiv), tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) (0.015 g, 0.016 mmol, 10 mol%) and 9,9-dimethyl-9H-xanthene-4,5-diylbis(diphenylphosphane) (XantPhos) (0.021 g, 0.037 mmol, 23 mol%) were added to a flask and suspended in toluene (4 mL) under a stream of N2. This mixture was stirred for 1 min before the premade acetic formic anhydride (75 μL, 0.95 mmol, 6 equiv) was added in a single portion, followed immediately by the addition of triethylamine (100 μL, 0.72 mmol, 4.5 equiv) while stirring. The flask was capped with a septum and stirred for 2 min. An additional portion of triethylamine was then added (100 μL) (1.44 mmol, 9 equiv total) followed by an additional portion of acetic formic anhydride (30 μL) (1.33 mmol, 8.3 equiv total). The reaction was stirred and heated to 85 °C for 18 h then cooled to 25 °C and diluted with ethyl acetate (5 mL). The crude reaction mixture was filtered through a small silica plug, concentrated in vacuo, and purified by silica column 1:99 MeOH:DCM–4:96 MeOH:DCM to give 0.012 g (16%) of an off-white solid: 1H NMR (CDCl3, 600 MHz) δ 9.76 (br s, 1H), 9.17 (s, 1H), 8.25 (dd, J = 8.4, 1.8 Hz, 1H), 8.09 (d, J = 7.8 Hz, 2H), 7.94 (d, J = 8.4 Hz, 1H), 7.65–7.43 (m, 5H), 7.35–7.75 (m, 3H), 3.25 (t, J = 7.8 Hz, 2H), 1.95 (quint, J = 7.8 Hz, 2H), 1.54 (app-sext, J = 7.8, 7.2 Hz, 2H), 1.03 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 174.4, 164.7, 164.4, 150.9, 145.4, 143.1, 140.3, 140.2, 134.5, 132.8, 132.4, 129.9, 129.6, 129.5, 128.9, 127.6, 126.1, 125.9, 124.7, 121.6, 34.0, 31.9, 22.3, 13.7. (ESI+): Calcd C28H24N3O3S [M + H] + 482.1533, found 482.1572 (error 8.1 ppm).
4-Amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline (3). 3h (0.010 g, 0.020 mmol) was added to a small vial and suspended in anhydrous methanol (3 mL). The mixture was sonicated for 1 min to break up any larger solid chunks. Under a stream of N2, concentrated sulfuric acid (3 drips, approximately 125 mg) was added, which quickly dissolved the fine suspended solids. The vial was capped and then stirred and heated at 60 °C for 15 h. The reaction was cooled to 25 °C and neutralized with solid sodium carbonate powder, diluted with ethyl acetate (5 mL) and water (5 mL), and stirred for 10 min. This mixture was concentrated in vacuo and redissolved in the same amount of ethyl acetate and water. The mixture was separated and the aqueous product was extracted with ethyl acetate (3 × 5 mL). The organics were combined and concentrated in vacuo. The crude product was purified by silica column 25:75 EtOAc:Hexs to give 4.5 mg (69%) of the final product 3 as a white waxy solid: 1H NMR (CDCl3, 600 MHz) δ 8.53 (d, J = 1.2 Hz, 1H), 8.03 (dd, J = 8.4, 1.2 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 6.86 (br s, 2H), 3.98 (s, 3H), 3.20 (t, J = 7.8 Hz, 2H), 1.91 (app-quint, J = 7.8, 7.2 Hz, 2H), 1.51 (sext, J = 7.2 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 174.5, 166.2, 151.0, 141.5, 138.7, 138.1, 131.5, 125.5, 125.2, 124.6, 121.5, 52.6, 33.9, 31.8, 22.2, 13.7. (APCI+): Calcd C16H18N3O2S [M + H] + 316.1114, found 316.1108 (error 1.9 ppm).



{kind=link}
{kind=link}
{kind=link}
{kind=link}


