
2,6-Bis[bis(1,1-dimethylethyl)phosphinito-κP]phenyl-κC]-trans-chlorohydro(phenylphosphine)iridium(III)



Abstract
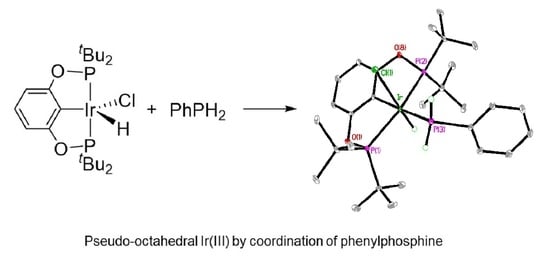
:
1. Introduction
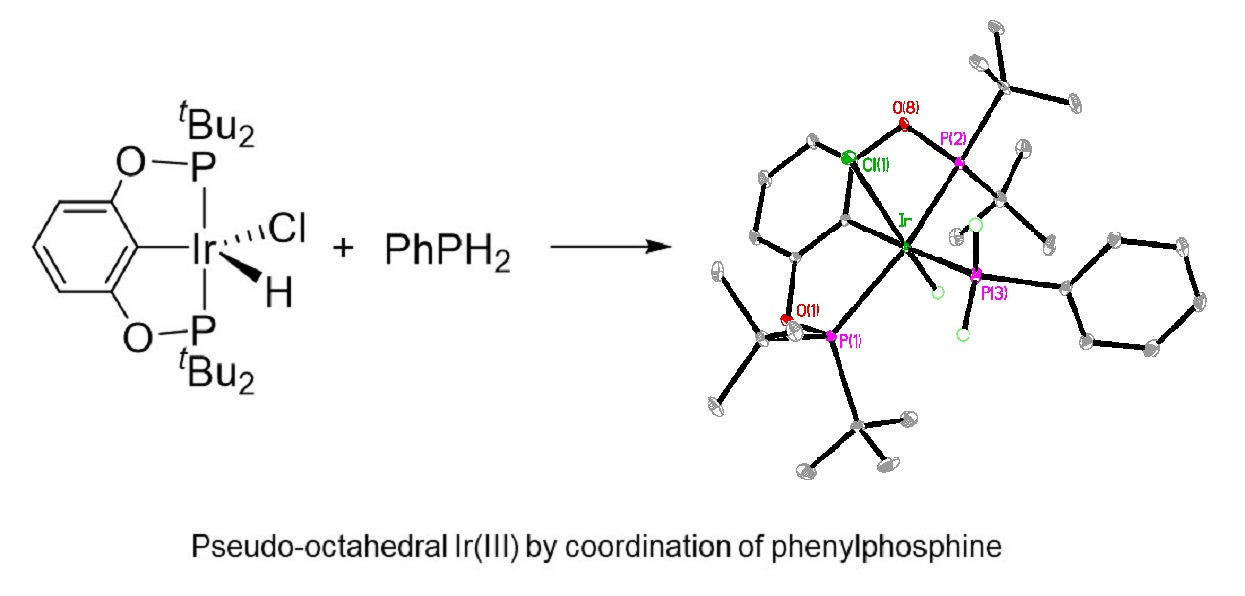
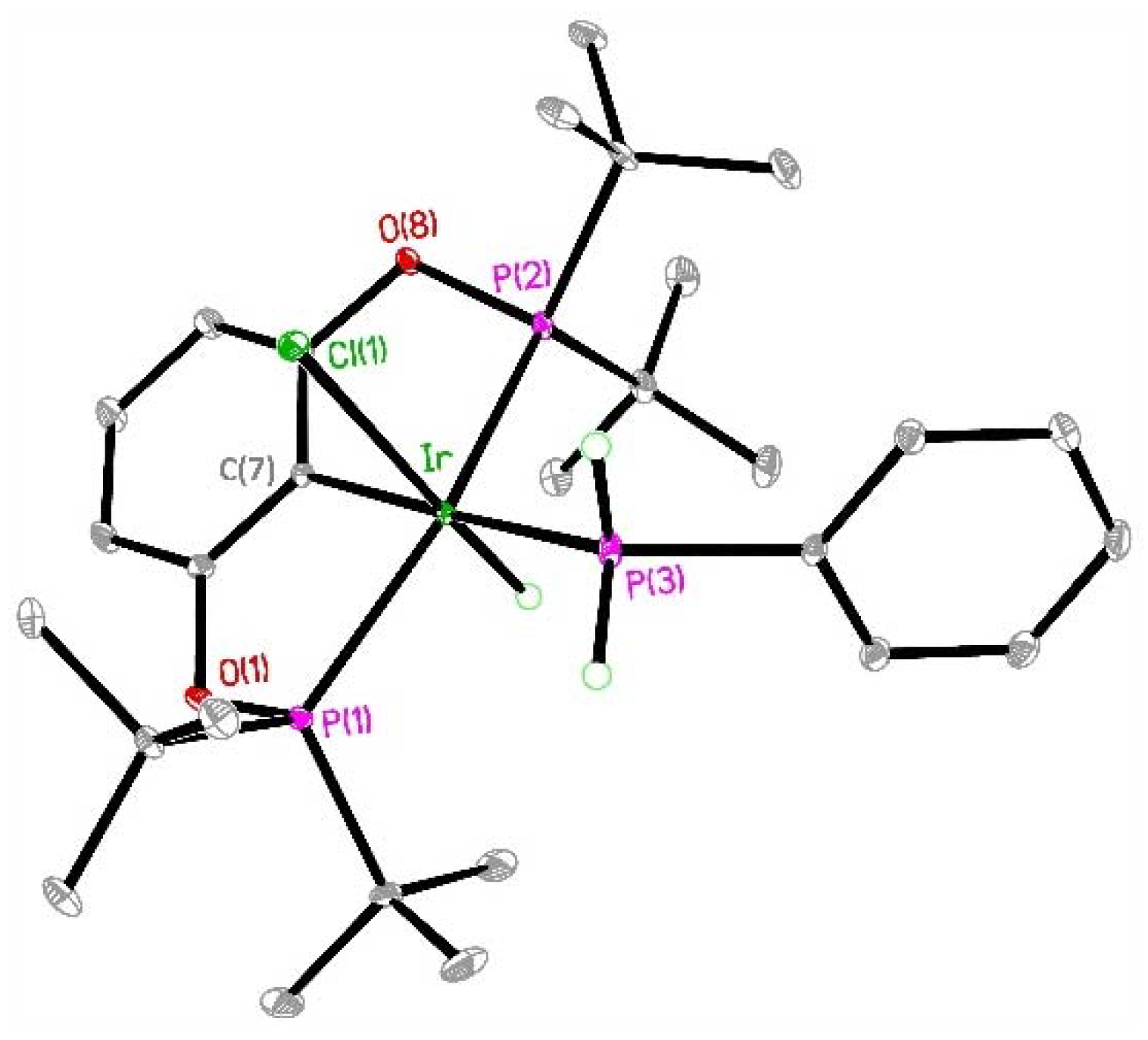
2. Results
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis of Compound 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Choi, J.; MacArthur, A.H.R.; Brookhart, M.; Goldman, A.S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rev. 2011, 111, 1761–1779. [Google Scholar] [CrossRef]
- Göttker-Schnetmann, I.; White, P.; Brookhart, M. Iridium Bis(phosphinite) p-XPCP Pincer Complexes: Highly Active Catalysts for the Transfer Dehydrogenation of Alkanes. J. Am. Chem. Soc. 2004, 126, 1804–1811. [Google Scholar] [CrossRef]
- Göttker-Schnetmann, I.; Brookhart, M. Mechanistic Studies of the Transfer Dehydrogenation of Cyclooctane Catalyzed by Iridium Bis(phosphinite) p-XPCP Pincer Complexes. J. Am. Chem. Soc. 2004, 126, 9330–9338. [Google Scholar] [CrossRef]
- Morales-Morales, D.; Redón, R.o.; Yung, C.; Jensen, C.M. Dehydrogenation of alkanes catalyzed by an iridium phosphinito PCP pincer complex. Inorg. Chim. Acta 2004, 357, 2953–2956. [Google Scholar] [CrossRef]
- Mucha, N.T.; Waterman, R. Iridium Pincer Catalysts for Silane Dehydrocoupling: Ligand Effects on Selectivity and Activity. Organometallics 2015, 34, 3865–3872. [Google Scholar] [CrossRef]
- Dietrich, B.L.; Goldberg, K.I.; Heinekey, D.M.; Autrey, T.; Linehan, J.C. Iridium-Catalyzed Dehydrogenation of Substituted Amine Boranes: Kinetics, Thermodynamics, and Implications for Hydrogen Storage. Inorg. Chem. 2008, 47, 8583–8585. [Google Scholar] [CrossRef]
- Staubitz, A.; Sloan, M.E.; Robertson, A.P.M.; Friedrich, A.; Schneider, S.; Gates, P.J.; Schmedt, a.d.G.J.; Manners, I. Catalytic Dehydrocoupling/Dehydrogenation of N-Methylamine-Borane and Ammonia-Borane: Synthesis and Characterization of High Molecular Weight Polyaminoboranes. J. Am. Chem. Soc. 2010, 132, 13332–13345. [Google Scholar] [CrossRef] [PubMed]
- Staubitz, A.; Soto, A.P.; Manners, I. Iridium-catalyzed dehydrocoupling of primary amine-borane adducts: A route to high molecular weight polyaminoboranes, boron-nitrogen analogues of polyolefins. Angew. Chem. Int. Ed. 2008, 47, 6212–6215. [Google Scholar] [CrossRef] [PubMed]
- Paul, U.S.D.; Braunschweig, H.; Radius, U. Iridium-catalysed dehydrocoupling of aryl phosphine–borane adducts: Synthesis and characterisation of high molecular weight poly(phosphinoboranes). Chem. Commun. 2016, 52, 8573–8576. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R. Triamidoamine-Supported Zirconium Compounds in Main Group Bond-Formation Catalysis. Acc. Chem. Res. 2019, 52, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R. Dehydrogenative Bond-Forming Catalysis Involving Phosphines. Curr. Org. Chem. 2008, 12, 1322–1339. [Google Scholar] [CrossRef]
- Waterman, R. Metal-Phosphido and -Phosphinidene Complexes in P–E Bond-Forming Reactions. Dalton Trans. 2009, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R. Selective Dehydrocoupling of Phosphines by Triamidoamine Zirconium Catalysts. Organometallics 2007, 26, 2492–2494. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
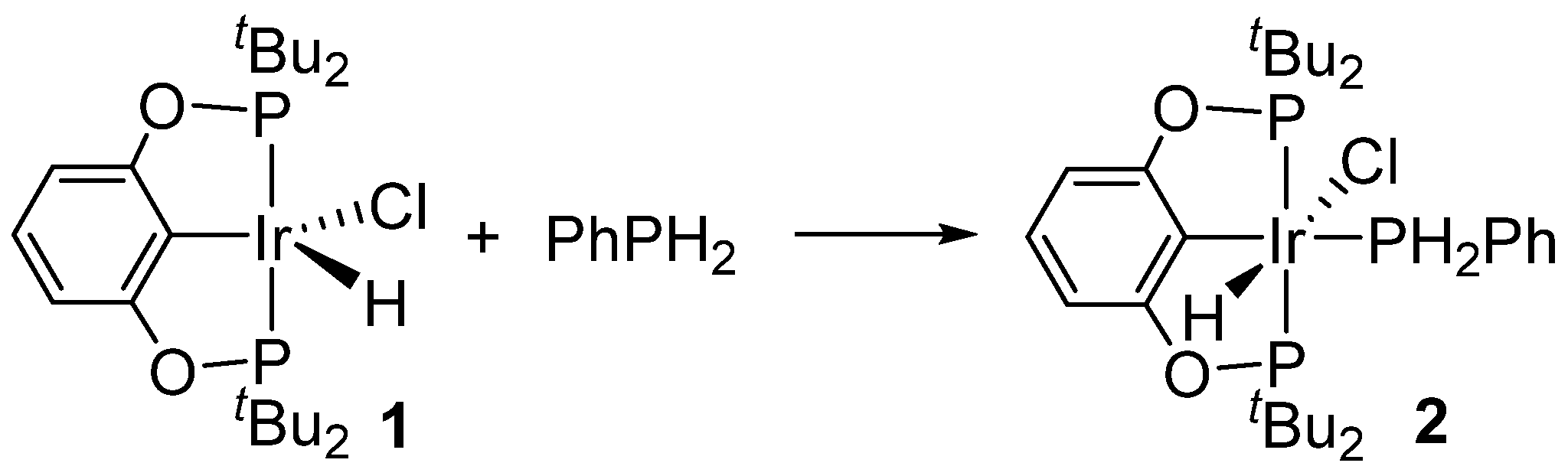{kind=link}
| Atom–Atom | Length [Å] | Atom–Atom–Atom | Angle [°] |
|---|---|---|---|
| Ir–C(7) | 2.0562(18) | P(3)–Ir–P(1) | 101.272(18) |
| Ir–P(3) | 2.3246(5) | P(3)–Ir–P(2) | 103.980(19) |
| Ir–P(1) | 2.3246(5) | P(3)–Ir–Cl(1) | 86.923(19) |
| Ir–P(2) | 2.3344(5) | P(1)–Ir–P(2) | 152.967(17) |
| Ir–Cl(1) | 2.4956(5) | C(7)–Ir–Cl(1) | 87.57(5) |
| Ir–H1 | 1.41(2) 1 | P(1)–Ir–Cl(1) | 97.438(18) |
| P(2)–Ir–Cl(1) | 93.415(18) | ||
| C(7)–Ir–P(3) | 174.40(5) | ||
| C(7)–Ir–P(1) | 78.47(5) | ||
| C(7)–Ir–P(2) | 77.34(5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucha, N.T.; Waterman, R. 2,6-Bis[bis(1,1-dimethylethyl)phosphinito-κP]phenyl-κC]-trans-chlorohydro(phenylphosphine)iridium(III). Molbank 2022, 2022, M1388. https://doi.org/10.3390/M1388
Mucha NT, Waterman R. 2,6-Bis[bis(1,1-dimethylethyl)phosphinito-κP]phenyl-κC]-trans-chlorohydro(phenylphosphine)iridium(III). Molbank. 2022; 2022(2):M1388. https://doi.org/10.3390/M1388
Chicago/Turabian StyleMucha, Neil T., and Rory Waterman. 2022. "2,6-Bis[bis(1,1-dimethylethyl)phosphinito-κP]phenyl-κC]-trans-chlorohydro(phenylphosphine)iridium(III)" Molbank 2022, no. 2: M1388. https://doi.org/10.3390/M1388
APA StyleMucha, N. T., & Waterman, R. (2022). 2,6-Bis[bis(1,1-dimethylethyl)phosphinito-κP]phenyl-κC]-trans-chlorohydro(phenylphosphine)iridium(III). Molbank, 2022(2), M1388. https://doi.org/10.3390/M1388