1. Introduction
Agmatine is a ubiquitous compound, discovered by Albrecht Kosse in herring sperm [
1]. It is a polyamine synthesized from the decarboxylation of L-arginine catalyzed by arginine decarboxylase. Until 1994, it was believed that agmatine was found only in bacteria, plants, and invertebrates, but then subsequent studies confirmed that agmatine is also expressed in mammals [
2]. Recent reports also demonstrated agmatine as a novel neurotransmitter [
3]. Ever since its discovery in humans, extensive research has been done to investigate its metabolic role, and that of the ensuing metabolites. Agmatine is known for its anti-depressive, anti-nociceptive, anxiolytic, and anti-inflammatory properties, and has been found to regulate heart rate and blood pressure, which makes it a potential therapeutic agent for the treatment of hypertension, ischemia, diabetes, and other cardiovascular ailments [
4,
5,
6,
7,
8].
The biological importance of agmatine and its therapeutic properties highlight the importance of the simple and efficient synthesis of agmatine. There are a limited number of chemical routes for the production of agmatine, but several biotechnical techniques exist in the literature. Multiple enzymatic methods have been reported for the production of agmatine, including the use of immobilized arginine decarboxylase and substrate-specific protein engineering [
8,
9,
10]. Through a whole-cell approach, Zhang et al. implemented the fermentative production of agmatine by metabolic engineering in
Escherichia coli [
11]. A similar approach was reported by Yang and co-workers, where the team overexpressed arginine decarboxylase in
Cornybacterium crenatu [
12]. Despite several biotechnical methods to synthesize agmatine, there are only a handful of chemical methods available to access agmatine. The existing chemical/industrial method of agmatine synthesis involves the treatment of 1,4-diaminobutane and cyanamide. To our knowledge, there is no reported synthetic route to produce labelled agmatine. In this work, we report a novel synthetic route to access labelled agmatine. Given the biological importance of agmatine and that of its metabolites, an efficient route to access labelled agmatine is necessary to investigate the biochemical pathways that regulate agmatine and its metabolites’ production, and the pathways that agmatine might regulate. With this facile synthetic route, we anticipate that agmatine production on a larger scale can be implemented as well.
2. Results and Discussion
Metabolites are continuously produced and consumed by a variety of biochemical reactions and pathways. To address this pool of biochemical information, measurements of metabolites by metabolomics using NMR and mass spectroscopy have gained much attention. Metabolomics has gained traction in a vast number of applications in the basic sciences, biology, medicine, and translational research [
13].
To provide insights into the metabolism of agmatine, we developed a chemical route (
Scheme 1) to synthesize agmatine (
3) and labelled agmatine (
7). Initial attempts to synthesize agmatine using the existing literature method were unsuccessful [
9]. We found that when a mixture of 1,4-diaminobutane (
1) and labelled cyanamide (
5) was heated at 50 °C under the reported conditions, no product formation was observed [
9]. Even changing the reaction parameters, such as increasing the temperature to 100 °C and increasing the reaction time, or the addition of a mild base such as potassium carbonate, failed to yield compound
7.
Therefore, we sought to develop a new strategy for the facile generation of
7 that is based on a two-step approach. The first step involves the treatment of
N-Boc-1,4-butanediamine (
4) with labelled cyanamide (
5) to generate
N-Boc-protected agmatine, followed by deprotection with trifluoroacetic acid (TFA) to yield the desired compound
7 (
Scheme 1). The resulting compound (compound
7) was successfully characterized by
1H NMR,
13C NMR,
19F NMR, HSQC, and mass spectroscopy.
1H NMR of compound
7 (labelled agmatine) showed the desired peaks corresponding to agmatine.
13C NMR too supported the product formation, which was evident from the
13C labelled peak at 164 ppm along with other desired peaks (
Supplementary Materials).
13C NMR (no decoupling) spectra were recorded for cyanamide (
5) and labelled cyanamide (
7), which revealed two distinct multiplets at 120 ppm and 163 ppm for labelled cyanamide (compound
5), while unlabelled cyanamide (compound
2) appeared as a singlet at 117 ppm. Literature reports revealed that
15N-labelled cyanamide (compound
5) can exist in two resonance forms (
Figure 1) [
14,
15].
Therefore, the two distinct peaks observed in the
13C NMR spectra of compound
5 were consistent with this. Further
13C NMR of compounds
3 and
7 were recorded, which showed singlets for both the compounds. A difference in the compounds was evident when
13C spectra were recorded with no decoupling. We noticed that in the case of compound
7, guanidium (terminal) labelled carbon that appeared to be a singlet in the
13C NMR spectrum (with decoupling), it appeared as two distinct triplets, while no such splitting was observed in the case of the unlabelled derivative (
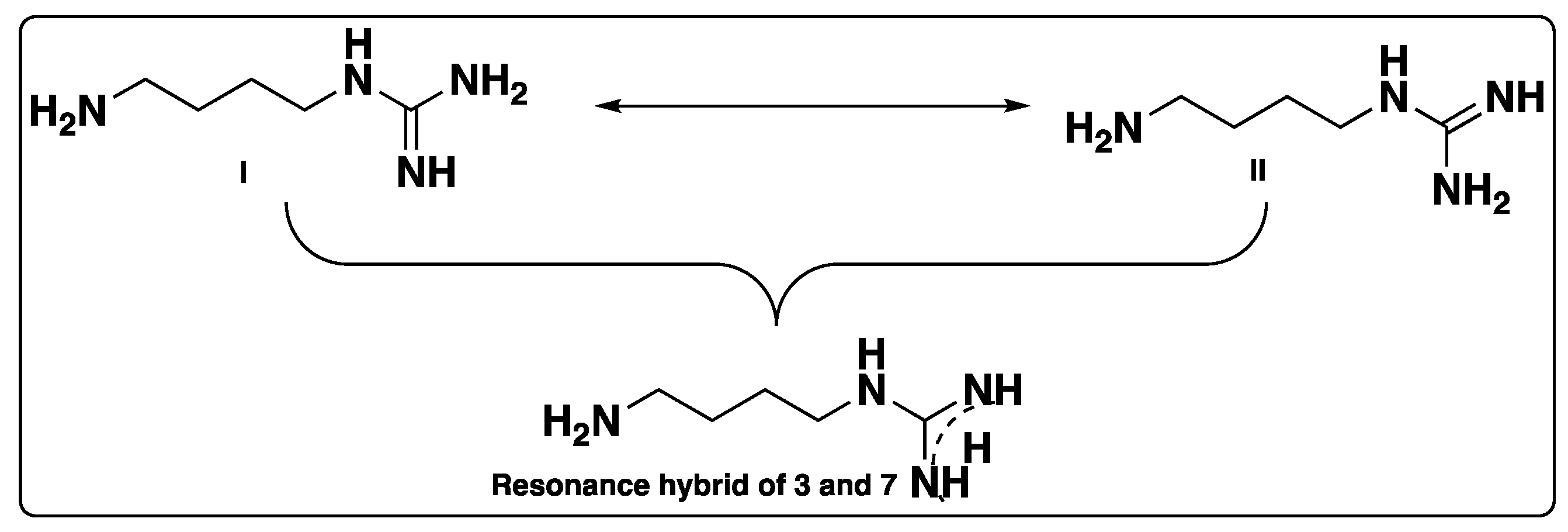
Supplementary Materials). Due to resonance or conjugation, compounds
3 and
7 can exist as form I or form II, and the delocalization can also be shown in the form of a resonance hybrid, as shown in
Figure 2. Structural identification of compound
7 was further supported by IR spectroscopy, which identified the presence of amine peaks at ~3200 cm
−1, and C=N and C-N peaks appeared at 1674 and 1200 cm
−1, respectively (
Supplementary Materials).
Further
1H-
15N HMBC confirmed the presence of
15N atoms. In addition, DEPT spectra were recorded to confirm the carbon skeleton. DEPT-135 of labelled agmatine (
7) showed a singlet around 160 ppm in the positive phase, while no such peak was observed in the case of the unlabelled derivative. The appearance of
13C in the positive phase in the DEPT-135 spectrum can be rationalized by the difference in the chemical environment (conjugation/resonance effects) imposed by two
15N atoms on the
13C atom in
7. In contrast, DEPT135 of compound
3 did not show a similar effect, and this was in agreement with the literature reports [
16].
Other 2D experiments such as COSY and HSQC helped in the structural identification of 7. Overall, the above experimental results support the unambiguous structural elucidation of labelled agmatine.
3. Methods and Reagents
All commercially available reagents and solvents were used without further purification. Reactions were monitored by both TLC and
1H NMR.
1H,
13C, and
19F NMR spectra were recorded on a Bruker Avance III HD 400 spectrometer at 400.11, 100.62, and 376 MHz, respectively, using the residual proton signal (
1H) for
1H NMR and the naturally abundant
13C carbon signal (
13C) of a deuterated solvent (DMSO, and CDCl
3) for
13C NMR as an internal standard. Chemical shifts are reported in the δ scale relative to residual CHCl
3 (7.26 ppm), D
2O (4.79), DMSO (2.5) for
1H NMR and for
13C NMR to the central line of CDCl
3 (77.16 ppm), DMSO (39.52).
1H NMR,
13C NMR (including DEPT,
13C NMR with no decoupling), and
19F NMR spectra were reported for the selected compounds and are included in the
Supplementary Materials. The following abbreviations have been used for the multiplicities: s = singlet, br = broad, d = doublet, and m = multiplet. Coupling constants (J) are reported in Hertz (Hz). Analytical TLCs were performed with Merck silica gel 60 F254 plates, and visualization of TLCs was accomplished under UV light. Mass spectra were obtained on an LTQ Orbitrap XL Mass Spectrometer (HESI source, positive polarity, capillary temp 200 °C, source voltage 3.0 kV). IR spectra were recorded using an FT-IR-4100, using potassium bromide as a supporting material (ATR method). HRMS and ESI spectra, along with the IR spectra, were recorded and reported for the selected compounds and can be found in the
Supplementary Materials. All solid reagents were weighed in an analytical balance without excluding moisture and air. Labelled cyanamide (cyanamide (stabilized with < 0.1% acetic acid) (
13C, 99%;
15N
2, 98%) 85%+ purity) was obtained from Cambridge Isotope Laboratories. Deionized water was used for all the aqueous reactions.
3.1. Synthesis of Tert-butyl N-[4-[(azanylcarbonimidoyl)amino]butyl]carbamate (6)
N-Boc-diaminobutane (4) (100 mg, 102 µL, 0.53) was added to labelled cyanamide (5) (20 mg, 0.44 mmoles) in anhydrous DMF (1 mL) and stirred to a clear solution. To this solution, diisopropylethylamine (74 mg, 100 µL, 0.57 mmoles) was added and the reaction mixture was heated to 80 °C for 24 h. Reaction progress was monitored by TLC. After completion, the reaction mixture was cooled to room temperature, water was added, and the resulting solution was extracted with ethyl acetate. The organic layer was separated, dried over magnesium sulfate, and, after filtration, the solvent was distilled under reduced pressure to yield a brown residue. The resulting crude compound was purified by flash column chromatography using a mixture of hexanes and ethyl acetate, and the pure intermediate (5) was isolated as a brown syrupy residue (yield: 46%).
1H NMR (400 MHz, CDCl3), δ, ppm: 1.29 (s, 9H, BOC-Hs), 1.42–1.54 (m, 4H, 2CH2), 2.98–3.11 (m, 2H, -CH2-NHBOC), 3.17–3.26 (m, 2H, -CH2NH), 4.85 (br s, -NH), 6.53 (br s, -NH); 13C NMR (101 MHz, CDCl3), δ, ppm: 26.5, 27.4, 37.6, 79.1, 156.1 (-C=O- of BOC), 161.5 (guanidium carbon).
3.2. Synthesis of 1-(4-Aminobutyl)guanidine-13C-2,3-15N2 (7)
To a solution of compound (5) (50 mg, 0.202 mmoles) in 2 mL of dichloromethane was added trifluoroacetic acid (TFA) (500 µL, 6.49 mmoles) at room temperature. The resulting solution was stirred at room temperature for 5 h. Once the reaction was complete, excess solvent and TFA was distilled off and co-distilled with dichloromethane twice. The resulting residue was dried under reduced pressure overnight to yield the labelled agmatine as a triflate salt (brown syrup, yield: 90%).
1H NMR (400 MHz, D2O), δ, ppm: 1.44–1.57 (m, 4H, 2CH2), 2.85–2.89 (t, 2H, -CH2NH, J = 14.2 Hz), 3.11–3.14 (t, 2H, -CH2NH, J = 13 Hz); 13C NMR (101 MHz, D2O), δ, ppm: 24.0, 25.2, 37.1, 38.9, 164.2 (13C-labelled carbon). (TFA peaks: 111.8–120.5 (q), 162.1–163.2 (q); 19F NMR (376 MHz, D2O), δ, ppm: −75.6.
1H NMR (400 MHz, DMSO), δ, ppm: 8.03 (br s, NH), 7.75 (br s, NH2), 3.06 (q, 2H, J = 6.3 Hz), 2.76 (t, 2H, J = 6.5 Hz), 1.4–1.51 (br m, 4H); IR (D2O) (KBr, v, cm−1): 3281 (NH), 3063 (NH), 2947 (CH), 1674 (C=N), 1202 (C-N), 1132 (C-N); HRMS (calculated mass: (M-NH2): 117.1649; observed mass (M-NH2): 117.1020.




{kind=link}
{kind=link}
{kind=link}


