Abstract
A series of benz[a]azulenes reacted with pyridinium hydrobromide perbromide (PHBPB) in hydrous tetrahydrofuran yielding the corresponding benz[a]azulenequinones in moderate yields. The detailed structure of the benz[a]azulenequinone derivative obtained was clarified by single crystal X-ray analysis.
1. Introduction
Isomeric abundance of azulenequinone has attracted the interest of many researchers in terms of their synthesis, structure, and reactivity, as well as theoretical aspects [1]. Morita and Takase attempted the preparation of the 2,6-azulenequinone derivative by the dehydrogenation of the corresponding 2,6-dihydroxyazulene derivative, but they were only able to isolate the dimeric form of the 2,6-azulenequinone derivative, owing to the high reactivity of the product [2]. For the synthesis of 1,2-azulenequinone derivatives, they have successfully prepared the derivatives from 2-acetoxyazulenes in a three-step method [3]. In 1984, Scott et al. reported the first synthesis of 1,4- and 1,6-azulenequinones (Figure 1) [4]. In the same year, they also accomplished the synthesis of 1,5- and 1,7-azulenequinones (Figure 1). In these syntheses, they first prepared the azulene diacetates in two steps from a readily available bicyclic trienone, and were further converted to the corresponding azulenequinones in three steps [5]. As a simpler method, Nozoe, Wakabayashi, and their co-workers demonstrated the reaction of parent azulene with bromine (Br2) in aqueous tetrahydrofuran (THF) to produce 1,5- and 1,7-azulenequinones as a mixture [6]. As the condensed derivatives, Fujimori, Yasunami, Nozoe et al. reported azulenequinones condensed with thiophene and furan, respectively, prepared by reacting the corresponding azulenothiophene and furan with Br2 in aqueous THF [7].
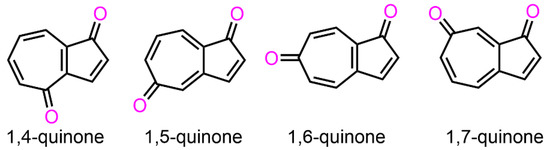
Figure 1.
A series of azulenequinones reported by Scott et al.
All the synthetic methods described above are useful for the preparation of azulenequinones, but these methods require a multi-step reaction or the use of a toxic reagent (i.e., Br2) that is difficult to handle. From this viewpoint, it is very important to develop a method for synthesizing the azulenequinones with the reagents that are easier to handle. In 2014, Sigrist and Hansen reported the reaction of benz[a]azulene with manganese dioxide (MnO2) to produce five benz[a]azulenequinone isomers (i.e., 5,10-, 6,10-, 7,10-, 8,10-, and 9,10-quinones) and fluorenone in very low yields [8]. However, their formation was only confirmed by TLC and mass spectrometry, and their structures were not determined by NMR spectroscopy and/or single crystal X-ray structure analysis. From this context, we describe herein a new synthetic method for the preparation of benz[a]azulenequinones from benz[a]azulenes, as well as their structural properties examined by NMR spectroscopic and X-ray crystallographic studies.
2. Results and Discussion
Pyridinium Hydrobromide Perbromide (PHBPB) shows similar reactivity to that of Br2 and is much easier to handle as it is a solid. Therefore, we explored the utilization of PHBPB as an oxidant for the preparation of azulenequinones. We investigated the oxidation of three-type benzo[a]azulene derivatives [9] with PHBPB to form benz[a]azulenequinones (Figure 2). The color of the aqueous THF solution of 1a immediately changed from blue to yellow upon the addition of PHBPB. After the general workup process, the reaction mixture was purified by silica gel chromatography to afford the benz[a]azulenequinone derivative 2a in 24% yield as the sole product (Table 1, entry 1). As described above, Sigrist and Hansen reported that the reaction of benz[a]azulene with MnO2 produces five types of benz[a]azulenequinones and fluorenone in very low yields. On the other hand, the formation of the sole product and higher yield of 2a may be attributed to the milder oxidizing nature of the PHBPB reagent compared to that of MnO2. Similar to the reaction of 1a, the reaction of 1b and 1c, which has an isopropyl group at the seven-membered ring, also proceeded to give the corresponding products 2b and 2c in moderate yields (2b, 42%; 2c, 43%). The higher yields of 2b and 2c compared to that of 2a might be attributed to the substitution of the i-Pr group at the seven-membered ring to improve the stability of the products.
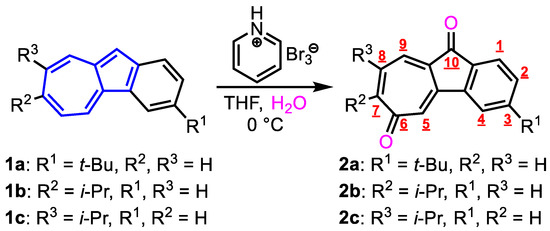
Figure 2.
The reaction of benz[a]azulenes 1a−1c with PHBPB.

Table 1.
Synthesis of benz[a]azulenequinones 2a−2c.
Benz[a]azulenequinones prepared in this study were characterized on the basis of spectral data, as shown in the “Materials and Methods”. High-resolution mass spectra of the reported compounds measured by the matrix-assisted laser desorption ionization-time of flight (MALDI−TOF) method showed the correct molecular ion peaks. The assignment of the proton signals observed in the 1H NMR spectra of these compounds was confirmed by COSY experiments. The 1H NMR spectra of 1b and 2b were shown in Figure 3. In compound 2b, the proton signals at 6,10-positions disappeared due to the oxidation at the C6 and C10 carbons. In addition, the signals attributed to two carbonyl carbons were found in the 13C NMR spectrum at δC = 186.2 and 190.9 ppm (see Supplementary Information). These results are consistent with the given structures of these products.
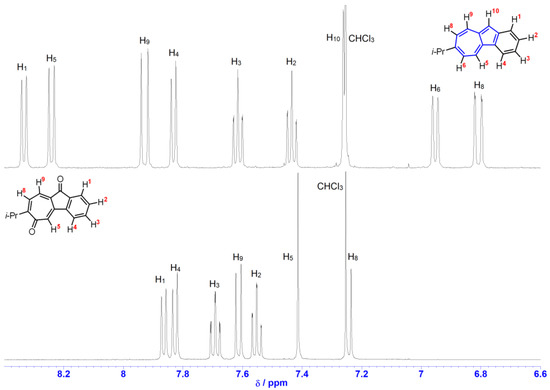
Figure 3.
1H NMR spectra of 1b (top) and 2b (bottom) in CDCl3 (500 MHz).
Since the single crystals were obtained from recrystallization in methanol, the X-ray structural analysis of 2a was performed. The crystal structure of 2a obtained by X-ray analysis and the respective bond lengths are shown in Figure 4. Even though the bond lengths of the fused benzene ring were almost the same as each other, alternating long and short bonds, i.e., bond alternation, were observed in the tropone (2,4,6-cycloheptatrien-1-one) moiety. Tropone is well known to exhibit aromatic properties based on the 6π-electron system arising from the contribution of the dipolar resonance structure induced by the polarization of the carbonyl group. On the other hand, the product 2a exhibited relatively large bond alternation, which indicates that the seven-membered ring may behave as non-aromatic cycloheptatriene rather than tropone, i.e., an aromatic ring. Furthermore, even though tropone is unstable and gradually decomposes, 2a−2c prepared in this study showed high stability that can be stored for several months under ambient conditions. Hence, 2a−2c may be served as 1-indenone derivatives rather than tropones.
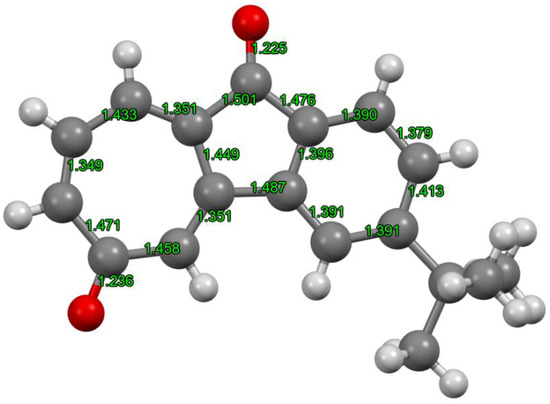
Figure 4.
X-ray structure and selected bond lengths of 2a (CCDC 1990090); recrystallized from methanol. Space group: P21/c, crystal system: monoclinic, a [Å] = 10.9399(6), b [Å] = 18.0026(8), c [Å] = 6.9786(4), α [°] = 90, β [°] = 105.291(6), γ [°] = 90, V [Å3] = 1325.76(13), Z value = 4, Dcalc [g/cm3] = 1.324, μ (MoKa) [mm−1] 0.085, R1 (I > 2.00σ) 0.0518, R1 (all reflections) 0.0733, wR2 (all reflections) 0.1326.
3. Materials and Methods
Melting points were determined with a Yanagimoto MPS3 micro-melting apparatus and were uncorrected. High-resolution mass spectra were obtained with a Bruker autoflex III TOF/TOF instrument (dithranol as a matrix substance and/or CF3CO2Ag as an auxiliary agent). IR spectra were measured with JASCO FT/IR-4100 spectrophotometer (JASCO Corporation, Tokyo, Japan). 1H and 13CNMR spectra were recorded in CDCl3 with a JEOL ECA500 (JEOL Ltd. Tokyo, Japan) at 500 MHz and 125 MHz, respectively. X-ray diffraction analysis was performed with a Rigaku VariMax with RAPID diffractometer (Rigaku Corporation, Tokyo, Japan) using multilayer mirror monochromated Cu Kα radiation (λ = 1.54187 Å) or by using a Rigaku VariMax with Saturn diffractometer using multilayer mirror monochromated Mo Kα radiation (λ = 0.71073 Å) at 100 ± 1 K. The crystals were mounted in cryoloops. Data collection was performed using RAPID AUTO or CrystalClear software (Rigaku Corporation, Tokyo, Japan). Data reduction was performed using RAPID AUTO, CrystalClear, or CrysAlisPro software. The data were corrected for Lorentz polarization and absorption effects. The structures were solved using SIR2004, SIR2011, or SHELXT 2018/2 and expanded using the Fourier technique. All calculations were performed using the Rigaku CrystalStructure crystallographic software package (Rigaku Corporation, Tokyo, Japan). SHELXL-2014/7 or SHELXL-2018/3 was used for structure refinement.
Synthesis of 2a: PHBPB (2.75 g, 8.59 mmol) was added at 0 °C to a solution of 1a (502 mg, 2.14 mmol) in THF (100 mL) and water (20 mL). The resulting mixture was stirred at the same temperature for 1.5 h. The reaction mixture was poured into water and extracted with CH2Cl2. The organic layer was washed with brine and dried with MgSO4. The crude product was purified by column chromatography on silica gel with toluene/AcOEt (1:1) as an eluent to give 2a (137 mg, 24%) as pale-yellow needles. M.p. 206−207 °C; IR (AT-IR): νmax = 3033 (w), 2966 (w), 2914 (w), 2870 (w), 1704 (s), 1638 (m), 1620 (w), 1595 (m), 1579 (s), 1557 (m), 1488 (w), 1464 (w), 1425 (m), 1397 (w), 1386 (m), 1367 (w), 1360 (m), 1334 (m), 1294 (w), 1284 (w), 1269 (w), 1240 (m), 1215 (m), 1194 (m), 1148 (w), 1125 (m), 1111 (m), 1081 (m), 1050 (w), 1033 (w), 979 (m), 957 (w), 921 (m), 896 (m), 887 (m), 846 (m), 813 (m), 784 (s), 743 (m), 681 (m), 632 (m), 615 (s), 602 (m), 571 (m), 542 (m), 526 (w), 506 (m), 482 (w), 461 (w), 449 (m), 418 (w), 403 (m) cm−1; 1H NMR (500 MHz, CDCl3): δH = 7.84 (d, J = 1.5 Hz, 1H, H-4), 7.82 (d, J = 8.0 Hz, 1H, H-1), 7.63 (dd, J = 8.0, 1.5 Hz, 1H, H-2), 7.56 (d, J = 7.8 Hz, 1H, H-9), 7.44 (dd, J = 2.6, 0.6 Hz, 1H, H-5), 7.20 (dd, J = 12.2, 7.8 Hz, 1H, H-8), 7.06 (dq, J = 12.2, 1.2 Hz, 1H, H-7), 1.39 (s, 9H, t-Bu) ppm; 13C{1H} NMR (125 MHz, CDCl3): δC = 190.11, 187.11, 160.85, 145.14, 145.12, 144.83, 140.10, 134.97, 133.28, 130.89, 129.89, 129.73, 124.81, 118.57, 77.37, 77.11, 76.86, 36.06, 31.15 ppm; HRMS (MALDI−TOF, positive) calcd for C18H16O2 + H+ [M + H]+ 265.1223, found 265.1238.
Synthesis of 2b: PHBPB (2.93 g, 9.16 mmol) was added at 0 °C to a solution of 1b (502 mg, 2.28 mmol) in THF (100 mL) and water (20 mL). The resulting mixture was stirred at the same temperature for 1.5 h. The reaction mixture was poured into water and extracted with CH2Cl2. The organic layer was washed with brine and dried with MgSO4. The crude product was purified by column chromatography on silica gel with toluene/AcOEt (1:1) as an eluent to give 2b (241 mg, 42%) as pale-yellow crystals. M.p. 107−108 °C; IR (AT-IR): νmax = 3067 (w), 2929 (w), 2871 (w), 1711 (s), 1644 (m), 1619 (w), 1600 (w), 1587 (m), 1573 (s), 1559 (s), 1469 (w), 1414 (m), 1380 (w), 1352 (w), 1335 (w), 1308 (m), 1266 (w), 1234 (m), 1197 (w), 1147 (w), 1127 (w), 1091 (w), 1061 (w), 1009 (s), 951 (w), 917 (s), 905 (m), 876 (m), 775 (s), 745 (m), 725 (s), 680 (w), 649 (m), 634 (m), 554 (m), 526 (w), 501 (m), 488 (s), 473 (m), 462 (m), 451 (w), 434 (m), 423 (m), 410 (s) cm−1; 1H NMR (500 MHz, CDCl3): δH = 7.87 (d, J = 7.6 Hz, 1H, H-1), 7.83 (d, J = 7.6 Hz, 1H, H-4), 7.69 (dd, J = 7.6, 1.0 Hz, 1H, H-3), 7.62 (d, J = 8.6 Hz, 1H, H-9), 7.55 (dd, J = 7.6, 1.0 Hz, 1H, H-2), 7.42 (s, 1H, H-5), 7.25 (d, J = 8.6 Hz, 1H, H-8), 3.53 (sept, J = 6.8 Hz, 1H, i-Pr), 1.21 (d, J = 6.8 Hz, 6H, i-Pr) ppm; 13C{1H} NMR (125 MHz, CDCl3): δC = 190.89, 186.16, 165.42, 144.86, 143.50, 137.23, 135.82, 135.74, 131.68, 130.85, 130.40, 128.75, 124.83, 121.76, 77.36, 77.10, 76.84, 31.09, 22.58 ppm; HRMS (MALDI−TOF, positive) calcd for C17H14O2 + H+ [M + H]+ 251.1067, found 251.1051.
Synthesis of 2c: PHBPB (5.75 g, 18.0 mmol) was added at 0 °C to a solution of 1c (867 mg, 3.94 mmol) in THF (175 mL) and water (35 mL). The resulting mixture was stirred at the same temperature for 1.5 h. The reaction mixture was poured into water and extracted with CH2Cl2. The organic layer was washed with brine and dried with MgSO4. The crude product was purified by column chromatography on silica gel with toluene/AcOEt (1:1) as an eluent to give 2c (420 mg, 43%) as pale-yellow crystals. M.p. 158−159 °C; IR (AT-IR): νmax = 3051 (w), 2964 (w), 2928 (w), 2872 (w), 1710 (s), 1638 (s), 1588 (s), 1574 (s), 1561 (s), 1461 (m), 1420 (m), 1381 (w), 1359 (w), 1335 (w), 1311 (m), 1288 (m), 1230 (m), 1189 (w), 1128 (w), 1101 (m), 1076 (m), 1024 (w), 994 (m), 953 (w), 915 (s), 897 (m), 878 (w), 843 (m), 800 (w), 765 (s), 725 (s), 699 (m), 652 (w), 595 (m), 567 (w), 547 (m), 519 (w), 503 (w), 493 (m), 480 (w), 469 (s), 449 (w), 434 (m), 412 (w), 405 (w) cm−1; 1H NMR (500 MHz, CDCl3): δH = 7.90 (d, J = 7.4 Hz, 1H, H-1), 7.83 (d, J = 7.7 Hz, 1H, H-4), 7.72 (t, J = 7.4 Hz, 1H, H-3), 7.57–7.60 (m, 2H, H-2,5), 7.38 (d, J = 2.6 Hz, 1H, H-9), 7.00 (br.s, 1H, H-7), 2.83 (sept, J = 6.8 Hz, 1H, i-Pr), 1.28 (d, J = 6.9 Hz, 6H, i-Pr) ppm; 13C{1H} NMR (125 MHz, CDCl3): δC = 190.78, 186.88, 155.62, 145.07, 143.93, 141.19, 138.10, 136.01, 135.44, 133.69, 131.96, 129.75, 125.00, 121.94, 38.32, 22.49 ppm; HRMS (MALDI−TOF, positive) calcd for C17H14O2 + H+ [M + H]+ 251.1067, found 251.1049.
4. Conclusions
In conclusion, we have described a new synthetic procedure for benz[a]azulenequinones from benz[a]azulenes. In this procedure, benz[a]azulenequinones were readily available by the oxidation of the corresponding benz[a]azulenes with PHBPB, which is easy to handle in moderate yields. The disappearance of two proton signals at the azulene moiety on the 1H NMR spectra and the formation of two carbonyl groups proved by 13C NMR spectra demonstrated the correctness of the structure of 2a−2c. Furthermore, single-crystal X-ray structure analysis revealed that the seven-membered ring of 2a exhibits pronounced bond alternation, suggesting the lower aromaticity of 2a at the tropone moiety rather than the tropone itself. Since the carbonyl group can be further functionalized by nucleophilic and condensation reactions, further transformations of 2a−2c are under investigation in our laboratory.
Supplementary Materials
The following supporting information can be downloaded online, Supplementary File S1: charts of 1H NMR, 13C NMR, COSY spectra and HRMS of compounds 2a−2c.
Author Contributions
Conceptualization, T.S. and M.Y.; methodology, A.Y. and N.S.; formal analysis, T.S., A.Y., N.S., R.S. and S.I.; investigation, T.S., A.Y., N.S., R.S. and S.I.; resources, T.S., A.Y., N.S. and M.Y.; writing—original draft preparation, T.S.; writing—review and editing, T.S., R.S. and S.I.; supervision, T.S.; funding acquisition, T.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number 21K05037.
Data Availability Statement
CCDC 1990090 (2a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scott, L.T.; Rozeboom, M.D.; Houk, K.N.; Fukunaga, T.; Lindner, H.J.; Hafner, K. The quinones of azulene. A theoretical prognosis. J. Am. Chem. Soc. 1980, 102, 5169–5176. [Google Scholar] [CrossRef]
- Morita, T.; Takase, K. The Synthesis of Diethyl 2,6-Dioxoazulene-1,3-Dicarboxylate, A Derivative of 2,6-Azulenedione (2,6-AzulenoQuinone). Chem. Lett. 1977, 6, 513–516. [Google Scholar] [CrossRef]
- Morita, T.; Karasawa, M.; Takase, K. The Synthesis and Some Properties of 1,2-Azulenequinone (1,2-Azulenedione) and its Derivatives. Chem. Lett. 1980, 9, 197–200. [Google Scholar] [CrossRef]
- Scott, L.T.; Griitter, P.; Chamberlain, R.E., III. Quinones of Azulene. 3. Generation and Trapping of the Reactive 1,4- and 1,6-Quinones. J. Am. Chem. Soc. 1984, 106, 4852–4856. [Google Scholar] [CrossRef]
- Scott, L.T.; Adams, C.M. Quinones of Azulene. 4. Synthesis and Characterization of the Parent 1,5- and 1,7-Quinones. J. Am. Chem. Soc. 1984, 106, 4857–4861. [Google Scholar] [CrossRef]
- Nozoe, T.; Wakabayashi, H.; Shindo, K.; Kurihara, T.; Ishikawa, S.; Kageyama, M. A Highly Convenient, One-Pot Synthesis of 3-Bromo-1,5- and -1,7-Azulenequinones by Polybromination of Azulene. Chem. Lett. 1995, 24, 25–26. [Google Scholar] [CrossRef]
- Matsuo, H.; Fujimori, K.; Ohta, A.; Kakehi, A.; Yasunami, M.; Nozoe, T. Synthesis of Azulenequinones Fused with Thiophene and Furan. Heterocycles 2003, 61, 271–279. [Google Scholar] [CrossRef]
- Sigrist, R.; Hansen, H.J. Benzo[a]azulenediones and 10,10′-Bibenzo[a]azulene. Helv. Chim. Acta 2014, 97, 1165–1175. [Google Scholar] [CrossRef]
- Shoji, T.; Yamazaki, A.; Katoh, R.; Shimamura, K.; Sakai, R.; Yasunami, M.; Okujima, T.; Ito, S. Synthesis, Reactivity, and Properties of Benz[a]azulenes via the [8 + 2] Cycloaddition of 2H-Cyclohepta[b]furan-2-ones with an Enamine. J. Org. Chem. 2022, 87, 5827–5845. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).