
Crystal Structures of Half-Sandwich Ru(II) Complexes, [(η6-p-Cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(X)]BF4, (X = Cl, Br, I)
 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
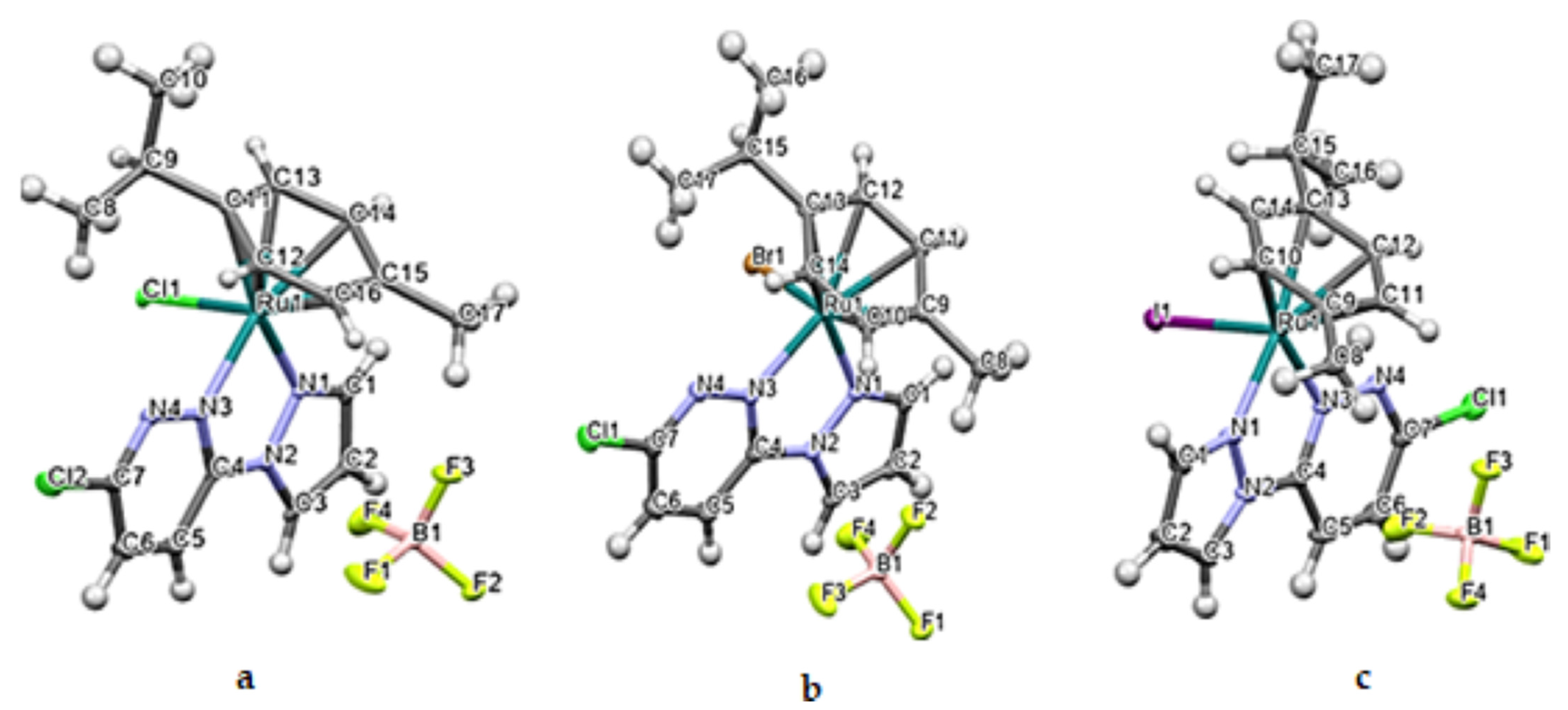
2.1. Crystal Structures of Ru1-3
2.2. Spectroscopic Data
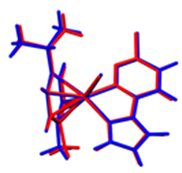
2.3. DFT-Calculated Optimized Structures
3. Materials and Methods
3.1. Reagents
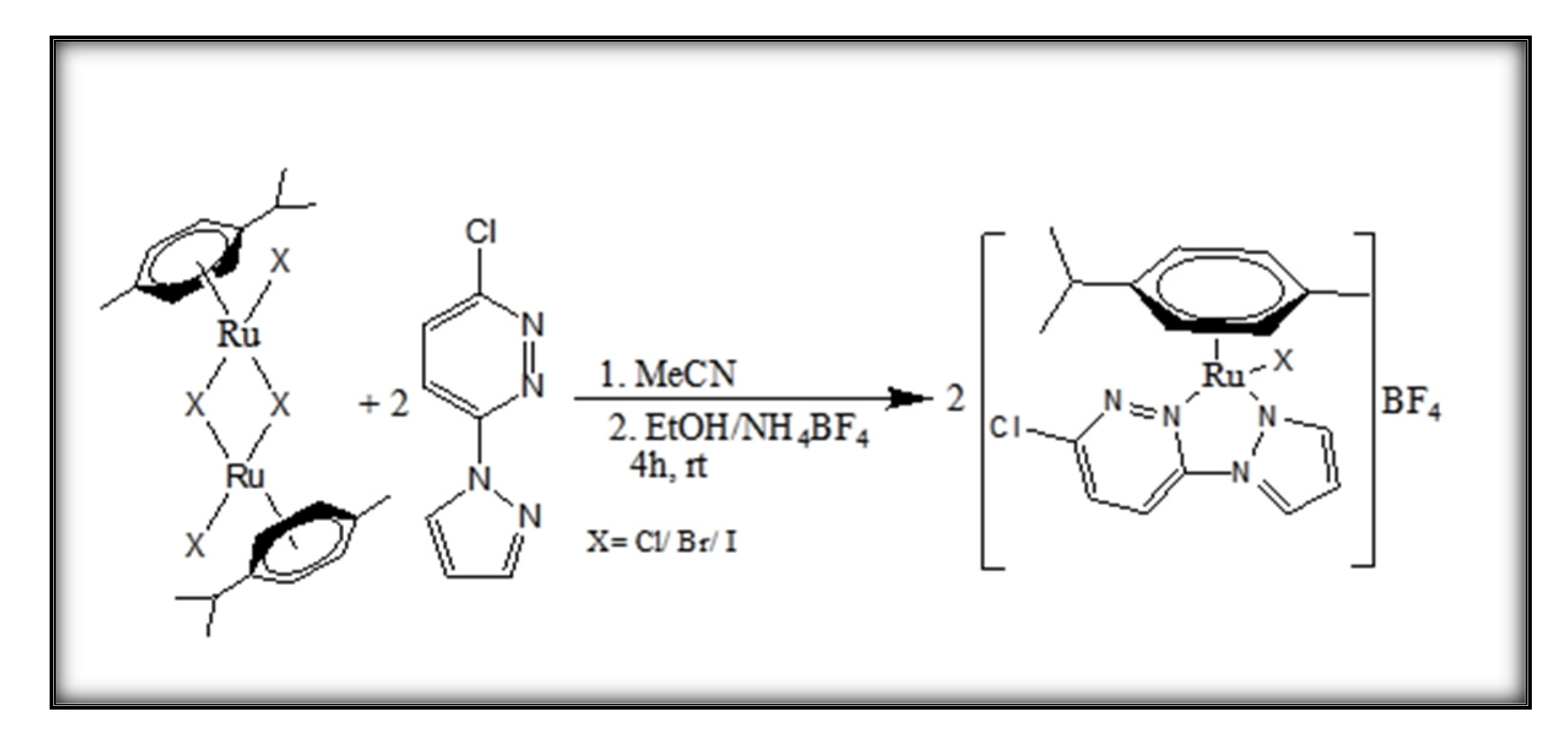
3.2. Synthesis of [(η6-p-cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(X)]BF4, Ru1 (X = Cl); Ru2 (X = Br); Ru3 (X = I)
3.3. Characterization of Complexes
3.4. DFT-Optimized Structures of Ru1-3
3.5. Listed Spectral Data of Ru1-3
- 3-chloro-6-(1H-pyrazol-1-yl)pyridazine (N,N′-L): White powder, 1H NMR (500 MHz, d6-DMSO) δ (ppm), 8.80 (d, J = 2.5 Hz, 1Hpzn), 8.27 (d, J = 9.3 Hz, 1Hpzn), 8.08 (d, J = 9.1 Hz, 1Hpdzn), 7.98 (d, J = 1.4 Hz, 1Hpdzn), 6.70 (dd, J = 4.3 Hz, 1Hpzn). 13C (100 MHz, d6-DMSO): δ (ppm) 154.1 (N-C-Npdzn), 153.7 (N=C-Cl, pdzn), 143.7 (C=N, pzn), 131.8 (C=Cpdzn), 127.8, (C=Npzn), 120.9 (C=Cpdzn), 109.7 (C=Cpzn) (see also Figure S14).
- [(η6-p-cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(Cl)]BF4 (Ru1): Yield 74.2%, orange crystals, MS (ESI)+ m/z (% Ipic) = 451 (100), [C17H19ClN4RuCl + H]+; 1H NMR (500 MHz, d6-DMSO) δ (ppm), 9.18 (d, J = 3.2 Hz, 1Hpzn), 9.02 (d, J = 2.2 Hz, 1Hpzn), 8.80 (d, J = 9.3 Hz, 1Hpdzn), 8.58 (d, J = 9.3 Hz, 1Hpdzn) 7.17 (dd, J = 3.2, 2.2 Hz, 1Hpzn), 6.17 (ddd, J = 12.1, 6.2, 1.1 Hz, 2HAr p-Cym), 5.99 (dd, J = 6.1, 1.1 Hz, 1H,Ar p-Cym), 5.87 (dd, J = 6.2, 1.1 Hz, 1Hp-Cym), 2.79 (p, J = 13.8, 6.9 Hz, 1Hp-Cym, CH(isopropyl), 2.12 (s, 3Hp-Cym, CH3), 1.15 (t, J = 6.9 Hz 6Hp-Cym, CH3(isopropyl). 13C (100 MHz, d6-DMSO): δ (ppm) 153.7 (N-C-Npdzn), 150.9 (C=Npzn), 148.8 (N=C-Clpdzn), 134 (C=Cpdzn), 132.9 (C=Npzn), 121.9 (C=Cpdzn), 112.8 (C=Cpzn), 105, 101.9, 86.7, 86.6, 84.2, 84.1 (CAr p-Cym), 30.3 Cp-Cym, CHisopropyl, 21.9 (-CH3p-Cym), 21.3, 17.8 (-CH3 isopropyl p-Cym). FTIR (KBr, cm−1 [(weak/medium/strong/sharp/broad = w/m/s/(shrp/br))]: 3085w(ν (Carom-H); 2959w, ν (C-Haliphatic), w(br); 1582, w(shrp), ν(N=Cpdzn/pzn); 1480, 1404, sshrp, ν(C=Ccym/pzn/,pdzn); 1284, m(shrp), ν(Ar βCHcym); 1050.37, vs(shrp), ν(BF4−); 820–850, 780, 650, mshrp ν[(Ru-Npdzn/pzn, Ru-Ccentroid cym, Ru-Cl], respectively. Elemental analysis: Calculated for [C17H19ClN4RuCl]BF4, %: C 37.94; H, 3.56; N, 10.41; found, % C, 37.82; H, 3.56; N, 10.50.
- [(η6-p-cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(Br)]BF4 (Ru2): Yield 75.5%, orange crystals, MS (ESI)+ m/z (% Ipic) = 497 (100), [C17H19ClN4RuBr + H]+; 1H NMR (500 MHz, d6-DMSO) δ (ppm), 9.18 (d, J = 3.2 Hz, 1Hpzn), 8.99 (d, J = 2.1 Hz, 1Hpzn), 8.78 (d, J = 9.3 Hz, 1Hpdzn, 8.57 (d, J = 9.3 Hz, 1Hpdzn), 7.17 (dd, J = 3.2, 2.1 Hz, 1Hpzn), 6.16 (m, 2HAr-p-Cym), 5.99 (m, 1Hp-Cym), 5.88 (dd, J = 6.3, 1.2 Hz, 1Hp-Cym), 2.86 (h, J = 6.9 Hz, 1H CHisopropyl p-Cym, 2.17 (s, 3H, p-Cym, CH3), 1.17 (dd, J = 8.6, 6.9 Hz, 6H, -CH3isopropyl p-Cym). 13C (100 MHz, d6-DMSO): δ (ppm) 153.6 (N-C-Npdzn), 150.8 (C=N, pzn), 149.1 (N=C-Cl, pdzn), 133.9 (C=Cpdzn), 132.9, (C=Npzn), 121.8 (C=Cpdzn), 112.9 (C=Cpzn), 105.4, 101.8, 86.6, 84.6, 84.5Ar, p-cym, 30.3 CHisopropyl), 21.9 (-CH3p-Cym), 21.3, 17.8, (-CH3isopropyl p-Cym). FTIR (KBr, cm−1, w/m/sshrp/br (= weak/medium/strongsharp/broad): 3083, w, ν(Carom-H); 2875-3, w, ν(C-Haliphatic), w(br); 1582, w(shrp), ν(N=Cpdzn/pzn); 1480, 1403, sshrp, ν(C=Ccym/pzn/,pdzn); 1168, m(shrp), ν(Ar βCHcym); 1021, vs(shrp), ν(BF4); 820–50, 780, 650, mshrp ν[(Ru-Npdzn/pzn, Ru-Ccentroid cym, Ru-Cl], respectively. Elemental analysis: Calculated for [C17H19ClN4RuBr]BF4, %: C 35.05; %H, 3.29; N, 9.62; found, % C, 35.72; H, 3.08; N, 9.48.
- [(η6-p-cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(I)]BF4 (Ru3): Yield 40%, orange crystals. MS (ESI)+ m/z (% Ipic) = 543 (100), [C17H19ClN4RuI + H]+; 1H NMR (500 MHz, d6-DMSO) δ (ppm), 9.19 (d, J = 3.2 Hz, 1Hpzn), 8.94 (d, J = 2.2 Hz, 1Hpzn), 8.78 (d, J = 9.3 Hz, 1Hpdzn, 8.55 (d, J = 9.3 Hz, 1Hpdzn), 7.17 (dd, J = 3.2, 2.2 Hz, 1Hpzn), 6.21 (dd, 1H, J = 6.4, 1.3 Hz), 6.14 (dd, J = 6.2, 1.2 Hz, 1Hp-Cym, 5.99 (dd, J = 6.4, 1.3 Hz, 1HAr-p-Cym), 5.94 (dd, J = 6.2, 1.2 Hz, 1Hp-Cym, 2.97 (h, J = 6.9 Hz, 1H, p-Cym, CHisopropyl, 2.21 (s, 3H, CH3 p-Cym), 1.19 (d, J = 11.0, 6.9 Hz, 6H, CH3isopropyl, p-Cym). 13C (100 MHz, d6-DMSO): δ (ppm) 153.5 (N-C-Npdzn), 150.3 (C=Npzn, 149.8 (N=C-Clpdzn), 133.7 (C=Cpdzn), 133 (C=Npzn), 121.5 (C=Cpdzn), 113.5 (C=Cpzn), 106.0, 102.2, 86.8, 86.5, 85.5, 84.9 (Arp-Cym), 31.1 (CH,isopropyl p-Cym, 22.8 (-CH3p-Cym, 19.1, 21.9, -CH3isopropyl p-Cym). FTIR (KBr, cm−1, w/m/sshrp/br (= weak/medium/strongsharp/broad), ν(funct. gr)): 3107, w, ν(Carom-H); 2976, w, ν(C-Haliphatic), w(br); 1581, w(shrp), ν(N=Cpdzn/pzn); 1433, sshrp, ν(C=Ccym/pzn/,pdzn); 1284, m(shrp), ν(Ar βCHcym); 1030, vs(shrp), ν(BF4); 820–50, 780, 650, mshrp ν[(Ru-Npdzn/pzn, Ru-Ccentroid. cym, Ru-Cl], respectively]. Elemental analysis: Calculated for [C17H19ClN4RuI]BF4, %: C 32.43; %H, 3.04; N, 8.90; found, % C, 32.92; H, 3.32; N, 9.07. 4.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gichumbi, J.M.; Friedrich, H.B. Half-sandwich complexes of platinum group metals (Ir, Rh, Ru and Os) and some recent biological and catalytic applications. J. Organomet. Chem. 2018, 866, 123–143. [Google Scholar] [CrossRef]
- Canivet, J.; Karmazin-Brelot, L.; Süss-Fink, G. Cationic arene ruthenium complexes containing chelating 1, 10-phenanthroline ligands. J. Organomet. Chem. 2005, 690, 3202–3211. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Ferri, M.G.; Hartinger, C.G.; Eichinger, R.E.; Stolyarova, N.; Severin, K.; Jakupec, M.A.; Nazarov, A.A.; Keppler, B.K. Influence of the spacer length on the in vitro anticancer activity of dinuclear Ruthenium—Arene compounds. Organometallics 2008, 27, 2405–2407. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Ferri, M.G.; Hartinger, C.G.; Nazarov, A.A.; Eichinger, R.E.; Jakupec, M.A.; Severin, K.; Keppler, B.K. Influence of the arene ligand, the number and type of metal centers, and the leaving group on the in vitro antitumor activity of polynuclear organometallic compounds. Organometallics 2009, 28, 6260–6265. [Google Scholar] [CrossRef] [Green Version]
- Matveevskaya, V.V.; Pavlov, D.I.; Samsonenko, D.G.; Ermakova, E.A.; Klyushova, L.S.; Baykov, S.V.; Boyarskiy, V.P.; Potapov, A.S. Synthesis and structural characterization of half-sandwich arene–ruthenium(II) Complexes with bis(imidazol-1-yl)methane, Imidazole and Benzimidazole. Inorganics 2021, 9, 34. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B.; Lazarus, G.G.; Singh, M.; Chenia, H.Y. Synthesis, characterization, anticancer and antimicrobial study of arene ruthenium(II) complexes with 1,2,4-triazole ligands containing an α-diimine moiety. Z. Naturforsch. B 2018, 73, 167–178. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; Van Den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [Green Version]
- Brindell, M.; Stawoska, I.; Supel, J.; Skoczowski, A.; Stochel, G.; van Eldik, R. The reduction of (ImH)[trans-Ru(III)Cl4(dmso)(Im)] under physiological conditions: Preferential reaction of the reduced complex with human serum albumin. JBIC 2008, 13, 909–918. [Google Scholar] [CrossRef]
- Mahmud, K.M.; Niloy, M.S.; Shakil, M.S.; Islam, M.A. Ruthenium Complexes: An alternative to platinum drugs in colorectal cancer treatment. Pharmaceutics 2021, 13, 1295. [Google Scholar] [CrossRef]
- Monro, S.; Colon, K.L.; Yin, H.; Roque III, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition metal complexes and photodynamic therapy from a tumor-centered approach: Challenges, opportunities, and highlights from the development of TLD1433. Chem. Rev. 2018, 119, 797–828. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Omondi, B.; Lazarus, G.; Singh, M.; Shaikh, N.; Chenia, H.Y.; Friedrich, H.B. Influence of halogen substitution in the ligand sphere on the antitumor and antibacterial activity of half-sandwich ruthenium(II) complexes [RuX(η6-arene)(C5H4N2-CH=N-Ar)]+. ZAAC 2017, 643, 699–711. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Samuelson, A.G. Substitution-modulated anticancer activity of half-sandwich ruthenium(II) complexes with heterocyclic ancillary ligands. Eur. J. Inorg. Chem 2014, 2014, 3536–3546. [Google Scholar] [CrossRef]
- Romero-Canelón, I.; Salassa, L.; Sadler, P.J. The contrasting activity of iodido versus chlorido ruthenium and osmium arene azo-and imino-pyridine anticancer complexes: Control of cell selectivity, cross-resistance, p53 dependence, and apoptosis pathway. J. Med. Chem. 2013, 56, 1291–1300. [Google Scholar] [CrossRef] [Green Version]
- Gumus, S. A computational study on substituted diazabenzenes. Turk. J. Chem. 2011, 35, 803–808. [Google Scholar] [CrossRef]
- Imran, M.; Asif, M. Study of various pyridazine and phthalazine drugs with diverse therapeutical and agrochemical activities. Russ. J. Bioorg. Chem. 2020, 46, 745–767. [Google Scholar] [CrossRef]
- Morris, R.E.; Aird, R.E.; del Socorro Murdoch, P.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef]
- Gupta, G.; Prasad, K.T.; Das, B.; Yap, G.P.; Rao, K.M. Ruthenium half-sandwich complexes with tautomerized pyrazolyl-pyridazine ligands: Synthesis, spectroscopic and molecular structural studies. J. Organomet. Chem. 2009, 694, 2618–2627. [Google Scholar] [CrossRef]
- Albertin, G.; Antoniutti, S.; Castro, J.; Garcí-Fontán, S. Preparation of pyrazole-pyrazolate half-sandwich complexes of ruthenium and osmium. Eur. J. Inorg. Chem 2011, 2011, 510–520. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B. Synthesis and characterization of half-sandwich ruthenium(II) complexes with N-alkylpyridyl-imine ligands and their application in transfer hydrogenation of ketones. Transit. Metal Chem. 2016, 41, 867–877. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B. Crystal structure of chlorido-(η6–1-isopropyl-4-methyl benzene)-(1-(pyridin-2-yl)-N-(p-tolyl)methanimine-κ2N,N)ruthenium(II) hexafluorophosphate(V), C23H26ClF6N2PRu. Z. Krist. New Cryst. Struct. 2017, 232, 285–287. [Google Scholar] [CrossRef]
- Neels, A.; Stoeckli-Evans, H.; Plasseraud, L.; Fidalgo, E.G.; Süss-Fink, G. Di-μ-bromo-bis [bromo(η6-para-cymene)ruthenium(II)] benzene solvate and di-μ-iodo-bis [(η6-para-cymene)iodoruthenium(II)] toluene solvate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1999, 55, 2030–2032. [Google Scholar] [CrossRef]
- Bacchi, A.; Cantoni, G.; Pelagatti, P. Polymorphs and co-crystal with half-sandwich Ru(II) dimers [(η6-arene) RuX 2]2. CrystEngComm 2013, 15, 6722–6728. [Google Scholar] [CrossRef]
- Wang, H.Y.; Qian, Y.; Wang, F.X.; Habtemariam, A.; Mao, Z.W.; Sadler, P.J.; Liu, H.K. Ruthenium(II)–Arene Metallacycles: Crystal Structures, Interaction with DNA, and Cytotoxicity. Eur. J. Inorg. Chem 2017, 2017, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.; Prasad, K.T.; Rao, A.V.; Geib, S.J.; Das, B.; Rao, K.M. Novel mononuclear η5-pentamethylcyclopentadienyl complexes of platinum group metals bearing pyrazolylpyridazine ligands: Syntheses and spectral studies. Inorg. Chim. Acta 2010, 363, 2287–2295. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B. Solvato-polymorph of [(η6-C6H6)RuCl (L)]PF6 (L=(2, 6-dimethyl-phenyl-pyridin-2-ylmethyleneamine). J. Mol. Struct. 2016, 1113, 55–59. [Google Scholar] [CrossRef]
- Desoize, B.; Madoulet, C. Particular aspects of platinum compounds used at present in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 317–325. [Google Scholar] [CrossRef]
- Wekesa, I.M.; Jaganyi, D. Kinetic and mechanistic studies of 1, 3-bis(2-pyridylimino)isoindolatePt(II) derivatives. Experimental and new computational approach. Dalton Trans. 2014, 43, 2549–2558. [Google Scholar] [CrossRef]
- Thompson, L.K.; Woon, T.; Murphy, D.B.; Gabe, E.J.; Lee, F.L.; Le Page, Y. Binuclear copper(II) complexes of a series of tetradentate pyrazolyldiazines. Crystal and molecular structures of [μ-3,6-bis (3,5-dimethyl-1-pyrazolyl)pyridazine-N,μ-N3,μ-N3,N](μ-hydroxo)dichlorodicopper(II)aquotrichlorocuprate hydrate, Cu3C14H21Cl5N6O3, and [μ-3,6-bis (3, 5-dimethyl-1-pyrazolyl)pyridazine-N,μ-N6,μ-N7, N](μ-hydroxo)tris(nitrato)diaquodicopper(II) hydrate, Cu2C14H23N9O13. Inorg. Chem. 1985, 24, 4719–4725. [Google Scholar] [CrossRef]
- CCD CrysAlis. CrysAlis Red; Xcalibur PX Software; Oxford Diffraction Ltd.: Abingdon, UK, 2008. [Google Scholar]
- Bruker APEX2. SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]
- Dolomanov, O.; Bourhis, L.; Gildea, R.; Howard, J.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | C17H19BCl2F4N4Ru | C17H19BBrClF4N4Ru | C17H19BClF4IN4Ru |
|---|---|---|---|
| M | 538.14 | 582.60 | 629.59 |
| T (K) | 104 | 100 | 102.6 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/n |
| Unit cell dimensions | |||
| a/Å | 14.0177(5) | 14.2238(3) | 14.5324(3) |
| b/Å | 10.6465(4) | 10.6770(2) | 10.7441(2) |
| c/Å | 14.9362(5) | 15.1270(3) | 15.1862(3) |
| α/° | 90 | 90 | 90 |
| β/° | 115.6560(10) | 117.1390(10) | 117.1050(10) |
| γ/° | 90 | 90 | 90 |
| Volume (Å3) | 2009.30(12) | 2044.37(7) | 2110.72(7) |
| Z | 4 | 4 | 4 |
| dcal g/cm3 | 1.779 | 1.893 | 1.981 |
|
Absorption coefficient (mm−1) | 9.206 | 10.136 | 19.085 |
| F(000) | 1072 | 1144 | 1216 |
| Crystal size (mm3) | 0.656 × 0.395 × 0.295 | 0.285 × 0.130 × 0.125 | 0.240 × 0.130 × 0.125 |
|
2θ range for data collection (°) | 6.996 to 142.948 | 6.984 to 135.992 | 6.98 to 136.082 |
| Index ranges | −16 ≤ h ≤ 16, −12 ≤ k ≤ 12, −18 ≤ l ≤ 17 | −17 ≤ h ≤ 16, −11 ≤ k ≤ 10, −18 ≤ l ≤ 15 | −17 ≤ h ≤ 17, −12 ≤ k ≤ 12, −18 ≤ l ≤ 18 |
| N(hkl), N(hkl)unique, Rint | 34776, 3815, 0.0419 | 25766, 3638, 0.0266 | 24963, 3808, 0.0318 |
|
Data/restraints/ Parameters | 3815/0/265 | 3638/0/265 | 3808/0/265 |
| Goodness-of-fit on F2 | 1.17 | 1.132 | 1.11 |
|
Final R indices [I ≥ 2σ (I)] | R1 = 0.0313, wR2 = 0.0794 | R1 = 0.0188, wR2 = 0.0469 | R1 = 0.0183, wR2 = 0.0424 |
|
Final R indices [all data] | R1 = 0.0313, wR2 = 0.0794 | R1 = 0.0190, wR2 = 0.0470 | R1 = 0.0186, wR2 = 0.0425 |
|
Largest diff. peak/hole/e Å−3 | 0.79/−1.58 | 0.55/−0.64 | 0.53/−0.72 |
| Complex | C17H19BCl2F4N4Ru | C17H19BBrClF4N4Ru | C17H19BClF4IN4Ru |
|---|---|---|---|
| Length (Å) | |||
| Ru1-‡X1 | 2.3920(6) | 2.5228(2) | 2.6996(2) |
| Ru1-N1pzn | 2.080(2) | 2.0718(17) | 2.073(2) |
| Ru1-N3pdzn | 2.079(2) | 2.0712(17) | 2.0743(19) |
| Ru-Cymcentroid | 1.676 | 1.676 | 1.679 |
| Angle (°) | |||
| N1-Ru1-X1 | 84.03(7) | 86.02(5) | 82.85(6) |
| N3-Ru1-X1 | 84.61(7) | 83.50(5) | 88.44(6) |
| N3-Ru1-N1 | 76.18(9) | 76.33(7) | 76.19(8) |
| Parameter | Ru1 | Ru2 | Ru3 |
|---|---|---|---|
| HOMO-LUMO energy/eV | |||
| −(LUMO), eV | 3.164 | 3.162 | 3.150 |
| −(HOMO), eV | 6.513 | 6.417 | 6.254 |
| Band gap, ΔE, eV | 3.349 | 3.255 | 3.104 |
| Global electrochemical parameter | |||
| Chemical hardness (η) | 1.675 | 1.627 | 1.552 |
| Chemical potential (μ) | −4.838 | −4.790 | −4.702 |
| Chemical softness (σ) | 0.597 | 0.614 | 0.644 |
| Electronegativity (χ) | 4.838 | 4.790 | 4.702 |
| Electrophilicity index (ω) | 6.988 | 7.048 | 7.122 |
| Nucleophilicity (ε) | 0.143 | 0.142 | 0.140 |
| Dipole moments (NBO) charge | 7.8607 | 8.5126 | 9.1474 |
| Ru | +0.090 | −0.057 | −0.130 |
| X | −0.295 | −0.213 | −0.110 |
| N1 | −0.184 | −0.186 | −0.188 |
| N3 | −0.211 | −0.213 | −0.215 |
| DFT-Optimized (Blue); Crystal Structure (Red) | HOMO | LUMO |
|---|---|---|
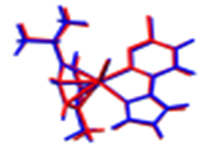 Ru1 |  | 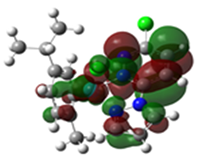 |
 Ru2 | 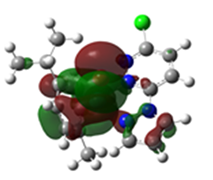 | 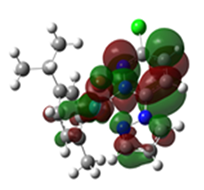 |
 Ru3 | 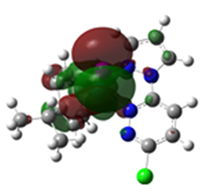 | 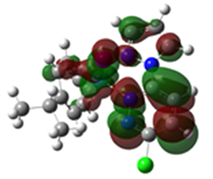 |
| Bond Lengths | Theoret. | Cryst. | %RE | Theoret. | Cryst | %RE | Theoret. | Cryst. | %RE |
|---|---|---|---|---|---|---|---|---|---|
| Ru-‡X | 2.3921 | 2.4529 | 2.5 | 2.5228 | 2.5982 | 2.9 | 2.6997 | 2.7644 | 6.5 |
| Ru-CCym(centroid) | 1.6760 | 1.8530 | 9.6 | 1.6760 | 1.8610 | 9.9 | 1.6790 | 1.8730 | 10.4 |
| Ru-N1 | 2.0789 | 2.0458 | 1.6 | 2.0719 | 2.0452 | 1.3 | 2.0711 | 2.0439 | 1.3 |
| Ru-N3 | 2.0802 | 2.0587 | 1.1 | 2.0743 | 2.0587 | 0.8 | 2.0731 | 2.0577 | 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mambanda, A.; Ongoma, P.; Gichumbi, J.; Omondi, R.O.; Hunter, L.A.; Kanyora, A.K. Crystal Structures of Half-Sandwich Ru(II) Complexes, [(η6-p-Cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(X)]BF4, (X = Cl, Br, I). Molbank 2022, 2022, M1477. https://doi.org/10.3390/M1477
Mambanda A, Ongoma P, Gichumbi J, Omondi RO, Hunter LA, Kanyora AK. Crystal Structures of Half-Sandwich Ru(II) Complexes, [(η6-p-Cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(X)]BF4, (X = Cl, Br, I). Molbank. 2022; 2022(4):M1477. https://doi.org/10.3390/M1477
Chicago/Turabian StyleMambanda, Allen, Peter Ongoma, Joel Gichumbi, Reinner O. Omondi, Leigh A. Hunter, and Amos K. Kanyora. 2022. "Crystal Structures of Half-Sandwich Ru(II) Complexes, [(η6-p-Cymene)(3-chloro-6-(1H-pyrazol-1-yl)pyridazine)Ru(X)]BF4, (X = Cl, Br, I)" Molbank 2022, no. 4: M1477. https://doi.org/10.3390/M1477