

(3-Methylene-2,3-dihydronaphtho[2,3-b][1,4]dioxin-2-yl)methanol


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
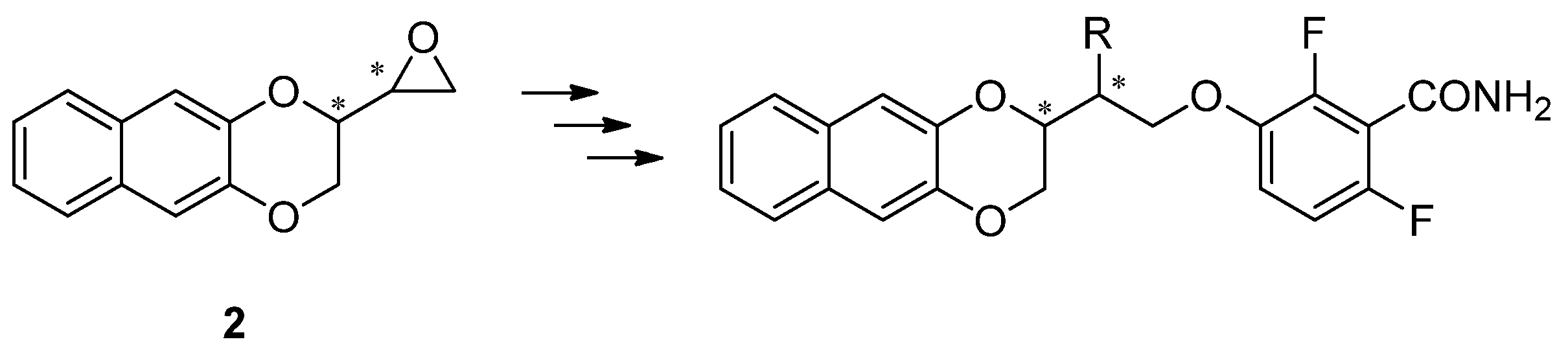
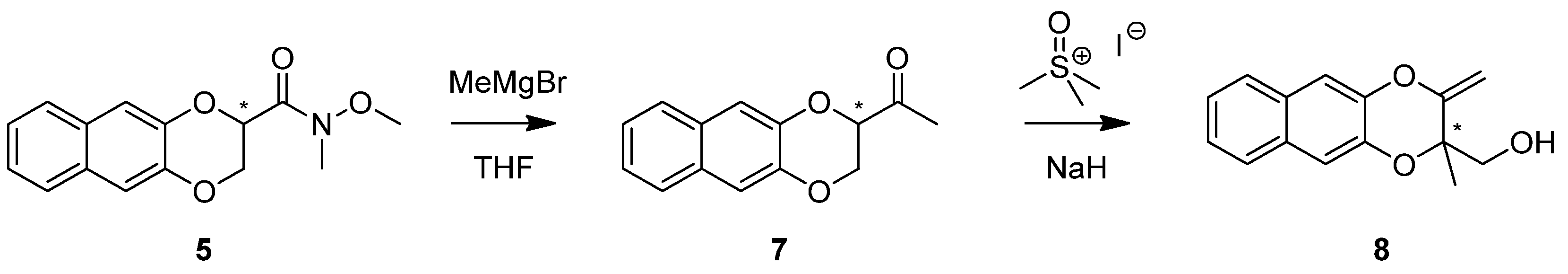
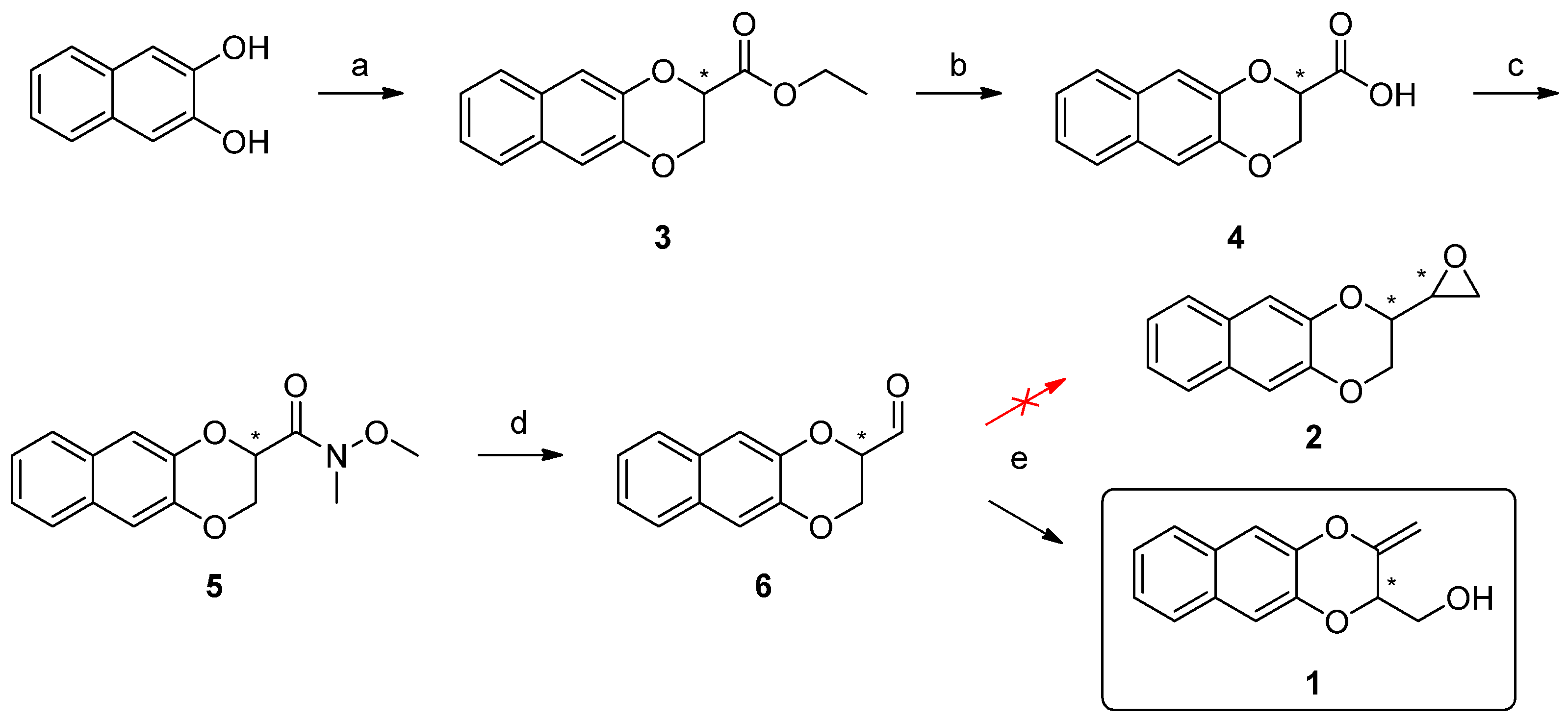
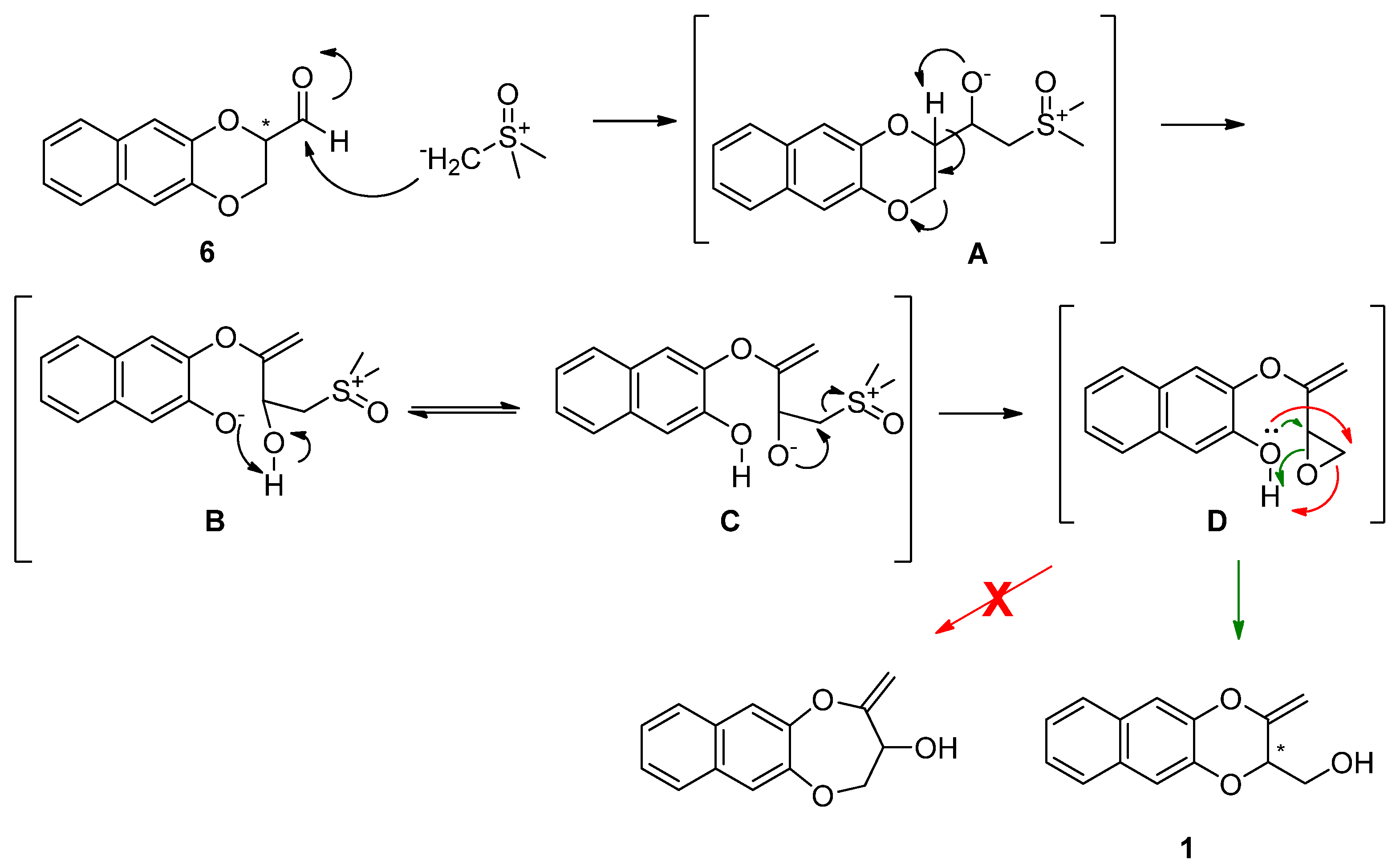
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethylsulfonium Methylide, a Reagent for Selective Oxirane Synthesis from Aldehydes and Ketones. J. Am. Chem. Soc. 1962, 84, 3782–3783. [Google Scholar] [CrossRef]
- Adamovskyi, M.I.; Artamonov, O.S.; Tymtsunik, A.V.; Grygorenko, O.O. The synthesis of a 2-azabicyclo [3.1.0]hexane by rearrangement of a spirocyclic epoxide. Tetrahedron Lett. 2014, 55, 5970–5972. [Google Scholar] [CrossRef]
- Kavanagh, S.A.; Piccinini, A.; Fleming, E.M.; Connon, S.J. Urea derivatives are highly active catalysts for the base-mediated generation of terminal epoxides from aldehydes and trimethylsulfonium iodide. Org. Biomol. Chem. 2008, 6, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-H.; Dai, L.-X.; Aggarwal, V.K. Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement. Chem. Rev. 1997, 97, 2341–2372. [Google Scholar] [CrossRef] [PubMed]
- Butova, E.D.; Fokin, A.A.; Schreiner, P.R. Beyond the corey reaction: One-step diolefination of cyclic ketones. J. Org. Chem. 2007, 72, 5689–5696. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Schwinden, M.D.; Radesca, L.; Patel, B.; Kronenthal, D.; Huang, M.-H.; Nugent, W.A. One-carbon chain extension of esters to alpha-chloroketones: A safer route without diazomethane. J. Org. Chem. 2004, 69, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Sebastián-Pérez, V.; Suigo, L.; Margolin, W.; Casiraghi, A.; Hrast, M.; Zanotto, C.; Zdovc, I.; Radaelli, A.; Valoti, E. Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket. Antibiotics 2021, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Casiraghi, A.; Fumagalli, L.; Valoti, E. How do reaction conditions affect the enantiopure synthesis of 2-substituted-1,4-benzodioxane derivatives? Chirality 2018, 30, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E. 2,6-Difluorobenzamide Inhibitors of Bacterial Cell Division Protein FtsZ: Design, Synthesis, and Structure-Activity Relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Artasensi, A.; Angeli, A.; Lammi, C.; Bollati, C.; Gervasoni, S.; Baron, G.; Matucci, R.; Supuran, C.T.; Vistoli, G.; Fumagalli, L. Discovery of a Potent and Highly Selective Dipeptidyl Peptidase IV and Carbonic Anhydrase Inhibitor as “Antidiabesity” Agents Based on Repurposing and Morphing of WB-4101. J. Med. Chem. 2022, 65, 13946–13966. [Google Scholar] [CrossRef] [PubMed]
- Clavier, S.; Khouili, M.; Bouyssou, P.; Coudert, G. Synthesis of naphtho [2,3-b][1,4]dioxin, 2-substituted naphtho [2,3-b][1,4]dioxins and 2,3-disubstituted naphtho [2,3-b][1,4]dioxins. Tetrahedron 2002, 58, 1533–1540. [Google Scholar] [CrossRef]
- Arrault, A. A Straightforward Synthesis of 1,2-Dihydronaphtho [2,1-b]furans from 2-Naphthols. Synthesis 1999, 1999, 1241–1245. [Google Scholar] [CrossRef]
- Howe, R.; Rao, B.S.; Chodnekar, M.S. Beta-adrenergic blocking agents. VII. 2-(1,4-benzodioxanyl) and 2-chromanyl analogs of pronethalol (2-isopropylamino-1-(2-naphthyl)ethanol). J. Med. Chem. 1970, 13, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Dominczak, N.; Lhoste, P.; Kryczka, B.; Sinou, D. Palladium-catalyzed heteroannulation of catechol with functionalized propargylic carbonates: Influence of the functional group on the regioselectivity of the cyclization. J. Mol. Catal. A Chem. 2007, 264, 110–118. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suigo, L.; Lodigiani, G.; Straniero, V.; Valoti, E. (3-Methylene-2,3-dihydronaphtho[2,3-b][1,4]dioxin-2-yl)methanol. Molbank 2022, 2022, M1521. https://doi.org/10.3390/M1521
Suigo L, Lodigiani G, Straniero V, Valoti E. (3-Methylene-2,3-dihydronaphtho[2,3-b][1,4]dioxin-2-yl)methanol. Molbank. 2022; 2022(4):M1521. https://doi.org/10.3390/M1521
Chicago/Turabian StyleSuigo, Lorenzo, Giulia Lodigiani, Valentina Straniero, and Ermanno Valoti. 2022. "(3-Methylene-2,3-dihydronaphtho[2,3-b][1,4]dioxin-2-yl)methanol" Molbank 2022, no. 4: M1521. https://doi.org/10.3390/M1521
APA StyleSuigo, L., Lodigiani, G., Straniero, V., & Valoti, E. (2022). (3-Methylene-2,3-dihydronaphtho[2,3-b][1,4]dioxin-2-yl)methanol. Molbank, 2022(4), M1521. https://doi.org/10.3390/M1521