
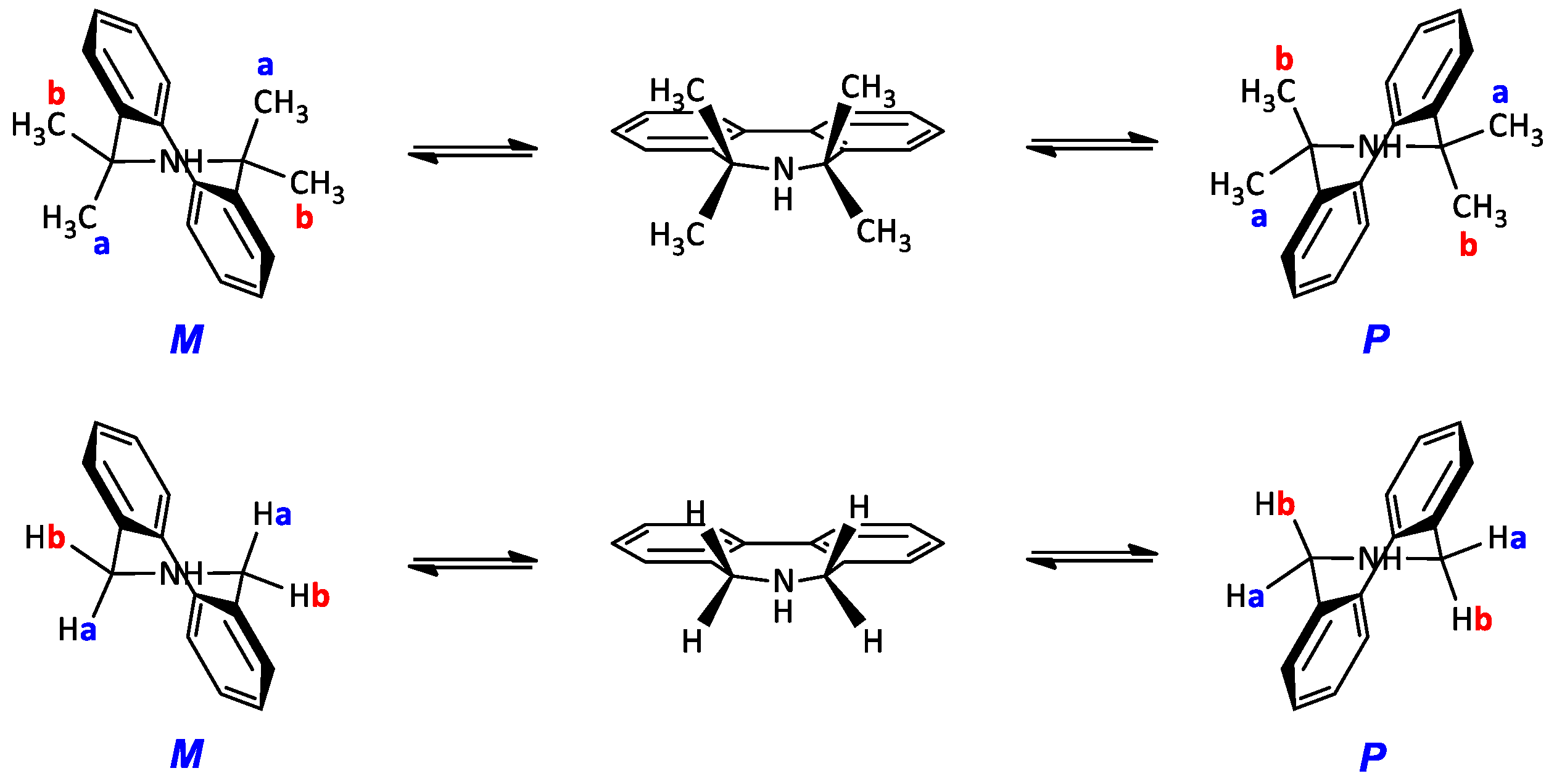
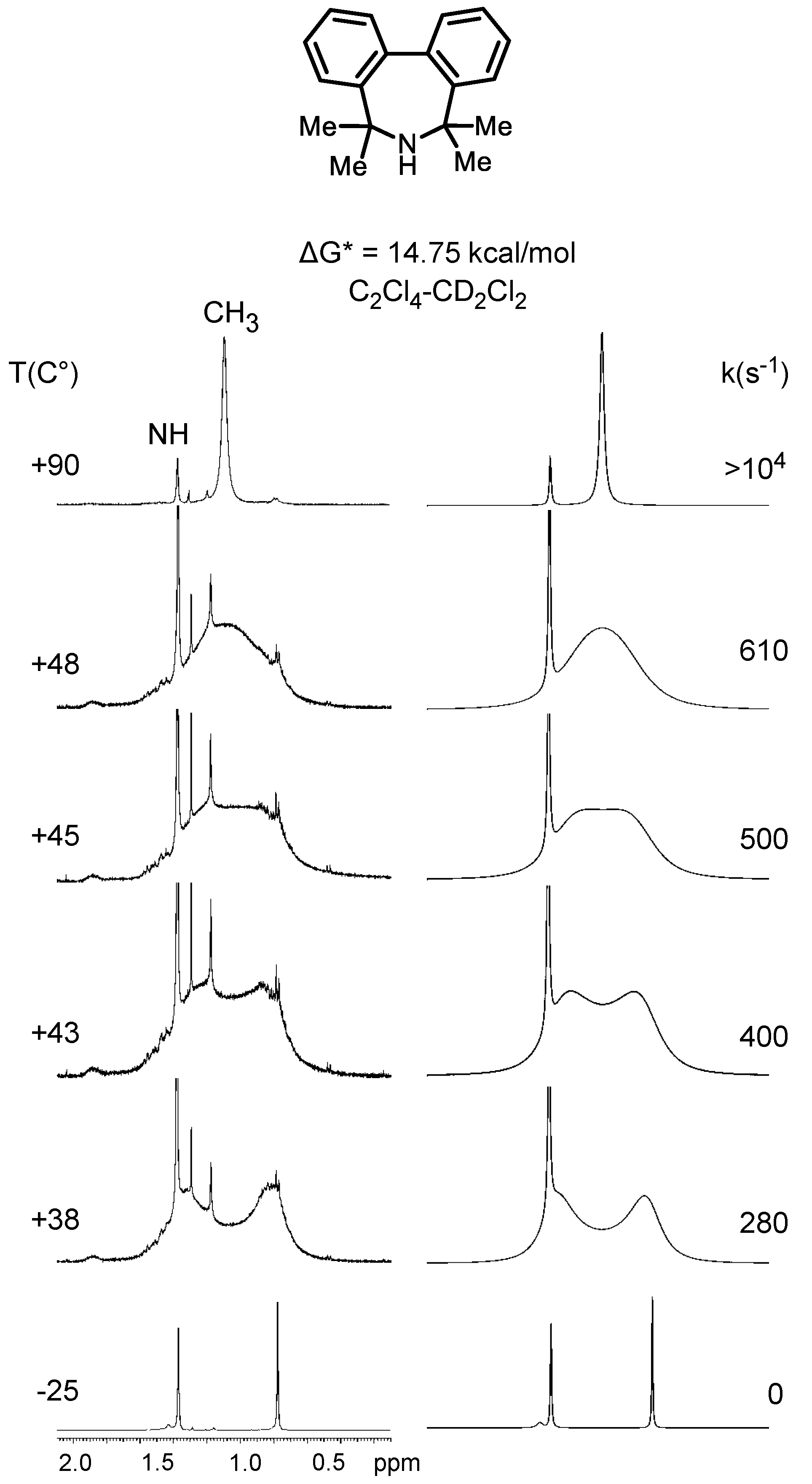
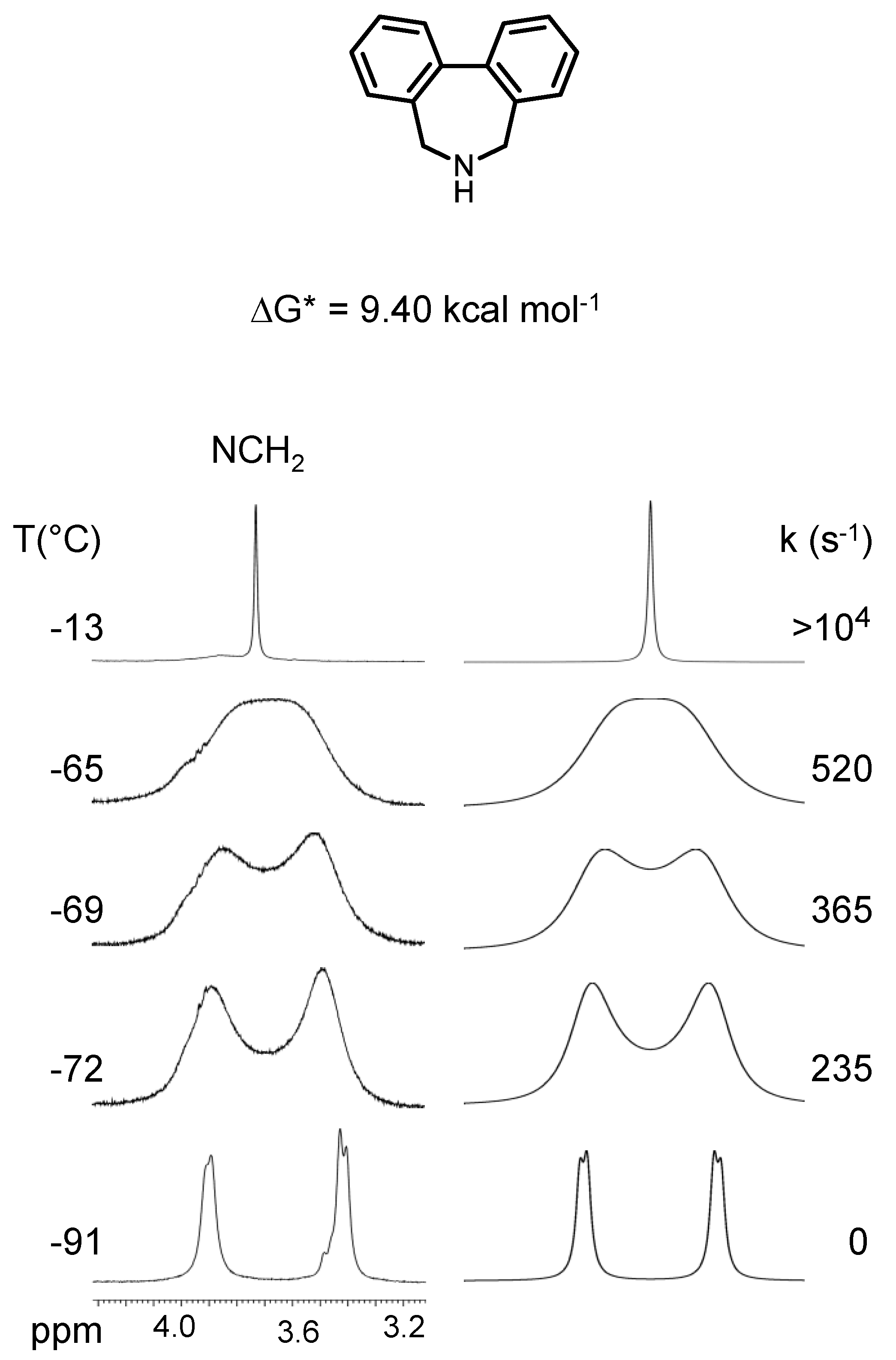
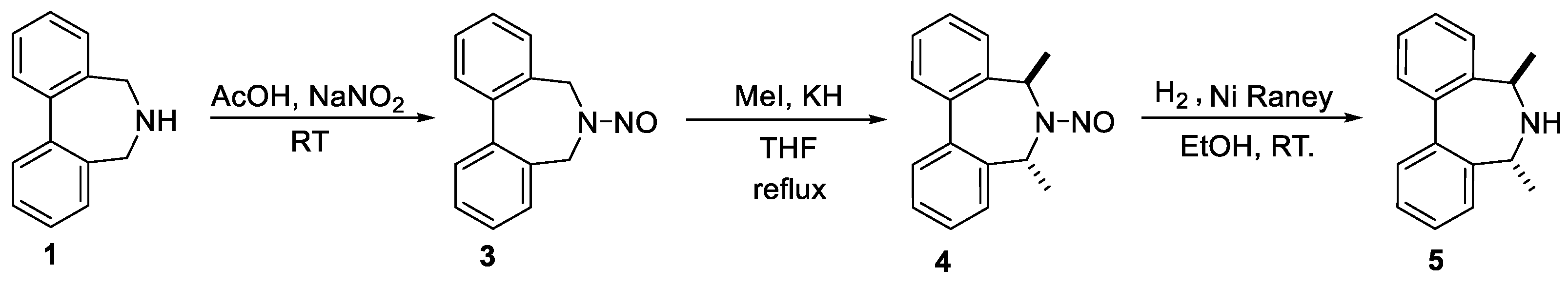
5,5,7,7-Tetrametyl-6,7-dihydro-5H-dibenzo[c,e]azepine



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
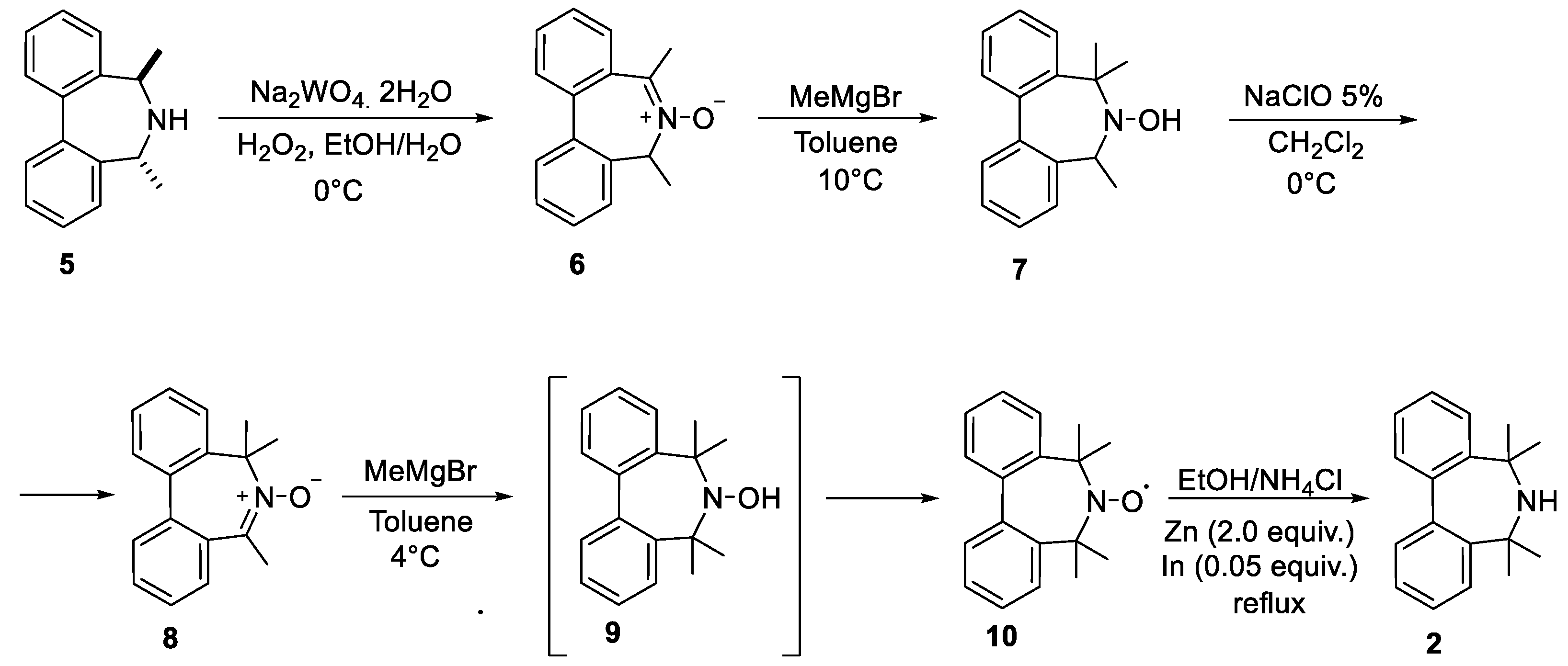
:1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mazaleyrat, J.P.; Cram, D.J. Chiral catalysis of additions of alkyllithiums to aldehydes. J. Am. Chem. Soc. 1981, 103, 4585–4586. [Google Scholar] [CrossRef]
- Superchi, S.; Mecca, T.; Giorgio, E.; Rosini, C. 1,1′-Binaphthylazepine-based ligands for asymmetric catalysis. Part 2: New aminoalcohols as chiral ligands in the enantioselective addition of ZnEt2 to aromatic aldehydes. Tetrahedron Asymmetry 2001, 12, 1235–1239. [Google Scholar] [CrossRef]
- Superchi, S.; Giorgio, E.; Scafato, P.; Rosini, C. Rational design of chiral 1,1′-binaphthylazepine-based ligands for the enantioselective addition of ZnEt2 to aromatic aldehydes. Tetrahedron Asymmetry 2002, 13, 1385–1391. [Google Scholar] [CrossRef]
- Pisani, L.; Superchi, S. 1,1′-Binaphthylazepine-based ligands for the enantioselective dialkylzinc addition to aromatic aldehydes. Tetrahedron Asymmetry 2008, 19, 1784–1789. [Google Scholar] [CrossRef]
- Pisani, L.; Superchi, S.; D’Elia, A.; Scafato, P.; Rosini, C. Synthetic approach toward cis-disubstituted γ- and δ-lactones through enantioselective dialkylzinc addition to aldehydes: Application to the synthesis of optically active flavors and fragrances. Tetrahedron 2012, 68, 5779–5784. [Google Scholar] [CrossRef]
- Kano, T.; Takai, J.; Tokuda, O.; Maruoka, K. Design of an axially chiral amino acid with a binaphthyl backbone as an organocatalyst for a direct asymmetric aldol reaction. Angew. Chem. Int. Ed. 2005, 44, 3055–3057. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Sugimoto, H.; Maruoka, K. Efficient organocatalytic cross-aldol reaction between aliphatic aldehydes through their functional differentiation. J. Am. Chem. Soc. 2011, 133, 18130–18133. [Google Scholar] [CrossRef]
- Summa, A.; Scafato, P.; Belviso, S.; Monaco, G.; Zanasi, R.; Longhi, G.; Abbate, S.; Superchi, S. Synthesis and stereochemical characterization of a novel chiral α-tetrazole binaphthylazepine organocatalyst. Molecules 2022, 27, 5113. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Kinsey, F.S.; Chan, Y.; Strutt, I.R.; Slawin, A.M.Z.; Jones, G.A. Novel binaphthyl and biphenyl α- and β-amino acids and esters: Organocatalysis of asymmetric diels–alder reactions. a combined synthetic and computational study. Org. Biomol. Chem. 2018, 16, 7400–7416. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Bartlett, C.J.; Chan, Y.; Day, D.; Parker, P.; Buckley, B.R.; Rassias, G.A.; Slawin, A.M.Z.; Allin, S.M.; Lacour, J.; et al. Asymmetric epoxidation using iminium salt organocatalysts featuring dynamically controlled atropoisomerism. J. Org. Chem. 2012, 77, 6128–6138. [Google Scholar] [CrossRef]
- Scafato, P.; Cunsolo, G.; Labano, S.; Rosini, C. Asymmetric activation of tropos catalysts in the stereoselective catalytic conjugate additions of R2Zn to α,β-enones: An efficient synthesis of (−)-muscone. Tetrahedron 2004, 60, 8801–8806. [Google Scholar] [CrossRef]
- Scafato, P.; Caprioli, F.; Rosini, C. Asymmetric addition of phenylboronic acid to cycloalkanones mediated by a rh/phosphoramidite complex: A comparison between tropos versus nontropos behavior. Tetrahedron Asymmetry 2011, 22, 558–561. [Google Scholar] [CrossRef]
- Superchi, S.; Marchitiello, V.; Pisani, L.; Scafato, P. Asymmetric addition of dimethylzinc to alkylidenmalonates mediated by phosphorous ligands: A new synthetic route to floral fragrances. Chirality 2011, 23, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Bochicchio, C.; Superchi, S.; Scafato, P. Tropos amino alcohol mediated enantioselective aryl transfer reactions to aromatic aldehydes: Enantioselective aryl transfer reactions to aromatic aldehydes. Eur. J. Org. Chem. 2014, 2014, 5939–5945. [Google Scholar] [CrossRef]
- Auras, S.; Trapp, O. Scorpio-ligand: Synthesis of biphenyl-dihydroazepine phosphoramidite ligands for asymmetric hydrogenation. Helv. Chim. Acta 2021, 104, e2100147. [Google Scholar] [CrossRef]
- Superchi, S.; Bisaccia, R.; Casarini, D.; Laurita, A.; Rosini, C. Flexible biphenyl chromophore as a circular dichroism probe for assignment of the absolute configuration of carboxylic acids. J. Am. Chem. Soc. 2006, 128, 6893–6902. [Google Scholar] [CrossRef]
- Vergura, S.; Scafato, P.; Belviso, S.; Superchi, S. Absolute configuration assignment from optical rotation data by means of biphenyl chiroptical probes. Chem. Eur. J. 2019, 25, 5682–5690. [Google Scholar] [CrossRef]
- Santoro, E.; Vergura, S.; Scafato, P.; Belviso, S.; Masi, M.; Evidente, A.; Superchi, S. Absolute configuration assignment to chiral natural products by biphenyl chiroptical probes: The case of the phytotoxins colletochlorin A and agropyrenol. J. Nat. Prod. 2020, 83, 1061–1068. [Google Scholar] [CrossRef]
- Vergura, S.; Orlando, S.; Scafato, P.; Belviso, S.; Superchi, S. Absolute configuration sensing of chiral aryl- and aryloxy-propionic acids by biphenyl chiroptical probes. Chemosensors 2021, 9, 154. [Google Scholar] [CrossRef]
- Vergura, S.; Pisani, L.; Scafato, P.; Casarini, D.; Superchi, S. Central-to-axial chirality induction in biphenyl chiroptical probes for the stereochemical characterization of chiral primary amines. Org. Biomol. Chem. 2018, 16, 555–565. [Google Scholar] [CrossRef]
- Saudan, L.A.; Bernardinelli, G.A.; Kündig, E.P. Diastereoselective Synthesis of (5R,7R)- and (5R,7S)-5,7-Dimethyl-6,7-Dihydro-5-H-Dibenz[c,e]Azepines. Synlett 2000, 2000, 483–486. [Google Scholar] [CrossRef]
- Pira, S.L.; Wallace, T.W.; Graham, J.P. Enantioselective route to 5-methyl- and 5,7-dimethyl-6,7-dihydro-5H -dibenz[c,e]azepine: Secondary amines with switchable axial chirality. Org. Lett. 2009, 11, 1663–1666. [Google Scholar] [CrossRef]
- Bottle, S.E.; Blinko, J.; George, G.; Micallef, A.; Wade, T. Profluorescent Nitroxide Compounds. WO Patent WO2007124543A1, 30 April 2007. [Google Scholar]
- Rychnovsky, S.D.; McLernon, T.L.; Rajapakse, H. Enantioselective oxidation of secondary alcohols using a chiral nitroxyl (n-oxoammonium salt) catalyst. J. Org. Chem. 1996, 61, 1194–1195. [Google Scholar] [CrossRef]
- Akhtar, M.H.; Oehschlager, A.C. The Decomposition of N-Arylazoamines. Tetrahedron 1970, 26, 3245–3263. [Google Scholar] [CrossRef]
- Cicchi, S.; Corsi, M.; Goti, A. Inexpensive and environmentally friendly oxidation of hydroxylamines to nitrones with bleach. J. Org. Chem. 1999, 64, 7243–7245. [Google Scholar] [CrossRef]
- Cicchi, S.; Bonanni, M.; Cardona, F.; Revuelta, J.; Goti, A. Indium-mediated reduction of hydroxylamines to amines. Org. Lett. 2003, 5, 1773–1776. [Google Scholar] [CrossRef] [PubMed]
- Gunther, H. NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry; Wiley-VCH: Chichester, UK, 2014; ISBN 9783527674770. [Google Scholar]
- Belviso, S.; Cammarota, F.; Rossano, R.; Lelj, F. Effect of polyfluorination on self-assembling and electronic properties of thioalkyl-porphyrazines. J. Porphyrins Phthalocyanines 2016, 20, 223–233. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisaccia, R.; Scafato, P.; Casarini, D.; Superchi, S. 5,5,7,7-Tetrametyl-6,7-dihydro-5H-dibenzo[c,e]azepine. Molbank 2023, 2023, M1554. https://doi.org/10.3390/M1554
Bisaccia R, Scafato P, Casarini D, Superchi S. 5,5,7,7-Tetrametyl-6,7-dihydro-5H-dibenzo[c,e]azepine. Molbank. 2023; 2023(1):M1554. https://doi.org/10.3390/M1554
Chicago/Turabian StyleBisaccia, Roberto, Patrizia Scafato, Daniele Casarini, and Stefano Superchi. 2023. "5,5,7,7-Tetrametyl-6,7-dihydro-5H-dibenzo[c,e]azepine" Molbank 2023, no. 1: M1554. https://doi.org/10.3390/M1554
APA StyleBisaccia, R., Scafato, P., Casarini, D., & Superchi, S. (2023). 5,5,7,7-Tetrametyl-6,7-dihydro-5H-dibenzo[c,e]azepine. Molbank, 2023(1), M1554. https://doi.org/10.3390/M1554