
Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate



Bioorganic Chemistry Laboratory, Facultad de Ciencias Básicas y Aplicadas, Universidad Militar Nueva Granada, Cajicá 250247, Colombia
*
Author to whom correspondence should be addressed.
Molbank 2024, 2024(1), M1794; https://doi.org/10.3390/M1794
Submission received: 15 February 2024
/
Revised: 15 March 2024
/
Accepted: 16 March 2024
/
Published: 19 March 2024
(This article belongs to the Section Organic Synthesis and Biosynthesis)
Abstract
:The multicomponent reaction between ʟ-tryptophan 1, 2-oxobutanoic acid 2, and 1-butanol in the presence of SiMe3Cl was studied using microwave irradiation conditions. The main product was identified as an unreported acetal-containing compound, namely, butyl (2,2-dibutoxybutanoyl)-ʟ-tryptophanate (3), yielding 89%. NMR experiments demonstrated that the adjacent methylene protons of the acetal group appeared as two signals exhibiting their behavior as diastereotopic protons. DFT/B3LYP calculations revealed an asymmetric molecular structure with specific angles, leading to an explanation of the NMR results. The calculated chemical shifts showed slight differences with the experimental values and suggested magnetic anisotropy and inductive deprotection around the methylene hydrogen atoms in the acetal location. The reaction mechanism was proposed in which SiMe3Cl plays a crucial role by promoting water removal through key steps.
1. Introduction
Acetals, also known as geminal-diethers, are organic compounds with a functional group R2C(OR′)2, where the R groups are usually organic fragments constituted by a carbon atom, with hydrocarbon chains attached to that or one hydrogen atom. In contrast, the R’ groups must be exclusive organic fragments [1]. These compounds can usually be formed from and also be convertible to aldehydes or ketones, having the same oxidation state at the central carbon but remarkably different chemical stability and reactivity compared to the analogous carbonyl compounds [2].
Several applications in the organic synthesis of these compounds have been reported, including forming carbon–carbon, carbon–nitrogen, carbon–oxygen, carbon–sulfur, and carbon–selenium bonds [3,4,5,6,7]. Kidjemet demonstrated using DMF·DMA mixtures to convert cis-diols to olefins, using some substrates such as diethyl ᴅ-tartrate [8]. Kinderman reported the palladium-catalyzed amidation of alkoxyallenes under basic conditions to access the 1-ethylquinozilidine structural fragment [9]. Dhakshinamoorthy described novel methodologies [10] involving acetals, such as the chemoselective synthesis of geminal diacetates from aldehydes using acetic anhydride and cerium (IV) ammonium nitrate (CAN). Lippur has reported the synthesis of (2S,2′S)-bimorpholine from (R,R)-tartaric acid ester acetal in six steps with 50% of the total yield, involving cyanide-catalyzed amidation and cyclization in a single step with p-toluenesulfonyl imidazole [11]. The derivatization of N,N′-dibenzylbimorpholine provided quaternary bimorpholinium salts used as chiral phase transfer alkylation catalysts. Juma reported the preparation of acetal-protected (2,4-dioxocyclohex-1-yl)acetic acid derivatives by the allylation of dilithiated 1,3-cyclohexane-1,3-diones, the protection of carbonyl groups, and the oxidation of the alkene precursor [12]. Polyacetals (and ketals) and polymers containing acetal bonds in the main chain or pendant chains have also been reported as an essential class of degradable polymers that can be considered for biomedical applications with orthopedic purposes and biocompatible and conjugated polymers with applications in drug formulation and medicine regenerative [13].
The synthesis of acetals involves aldehydes or ketones as precursors, which can be transformed into acetals or geminal dieters [14]. Several methods have used glycerol as a precursor, acetalized with carbonyl compounds using an iron catalyst FeCl3·6H2O, forming cyclic acetals [15]. N-acyl-N and/or O-acetals can be synthesized from aldehydes, amides, and alcohols via a two-step protocol [16]. Metal ions can catalyze the amide group formation and cleavage of C-O bonds [17]. Recently, methodologies combining acetal formation and amidation reactions were described for biologically active compounds [18]. Vincent reported two procedures for preparing N,N-dibenzyl formamidines from primary amines [19]. Tsotinis reported the synthesis of pyrrolo [1′,2′:1,2][1,4]diazepin [7,6-b]indol-5(6H)-one compounds [20]. Wu synthesized spiropyrans by condensing hemiacetal chromene esters with nucleophiles [21]. Luo described the development of a scalable synthetic route for the insecticide broflanilide [22].
Recently, the reactions between alkyl esters derived from 2-amino acids such as ʟ-tryptophan 1 and 2-ketocarboxylic acids were explored by the Bioorganic Chemistry laboratory [23]. The reaction yields decreased when high-molecular-weight alcohols were used. Microwave irradiation presumably promoted kinetically favored amidation reactions. Small alcohols, e.g., methanol and ethanol, induced a subsequent reaction, forming a hemiacetal compound. When using isopropanol, initial transesterification and amidation occurred, but carbonyl group attack is not favored, preventing the formation of the described hemiacetal. Considering the relevance of this class of compounds, the specificity of its synthesis [24], as well as its potential use as a precursor of various biologically active compounds [25], the synthesis of an unreported acetal-containing compound, i.e., butyl (2,2-dibutoxybutanoyl)-ʟ-tryptophanate 3, is described for the first time in the present study. In addition, the previously reported methodology [23] was revisited, and the influence of the temperature and the reaction time in a model conversion consisting of ʟ-tryptophan 1, 2-oxobutanoic acid 2, and 1-butanol as precursors was investigated, evaluating the role of SiMe3Cl as a catalyst under microwave (MW) irradiation. The results are discussed below.
2. Results and Discussion
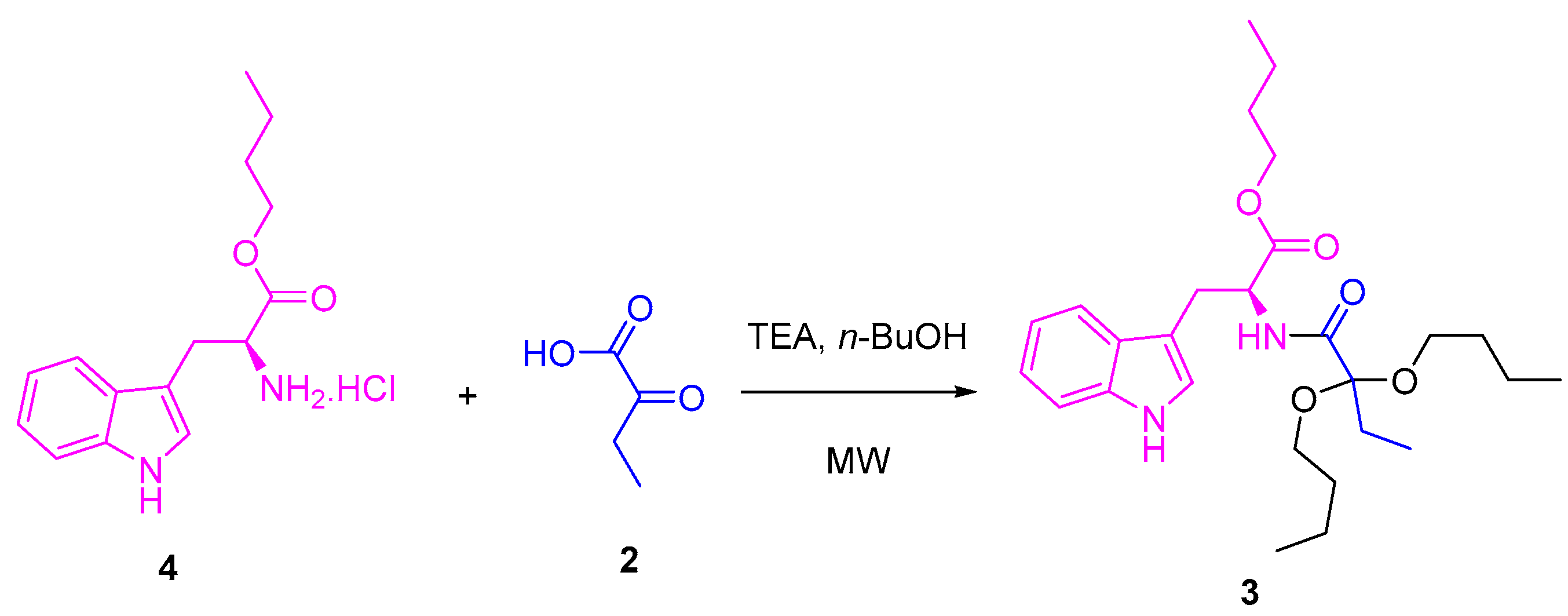
Several tests were carried out to achieve the synthesis of the title compound using ʟ-tryptophan 1 as a precursor. The method proposed by Quiroga [23] was initially used, whose ʟ-tryptophan butyl ester 4 [26] and 2-oxobutanoic acid 2 were used as precursors at the same stoichiometric ratio (1 eq:1 eq). These compounds were mixed with 1-butanol excess, and, after the addition of triethylamine, they were placed under microwave (MW) irradiation at several temperatures and times (Scheme 1, Table 1). Once the reactions ended, the target product was purified. In some cases, the product was identified as compound 3, with low yields between 5 and 22%. The spectroscopic data are presented in the Supplementary Materials (Figures S1–S3).
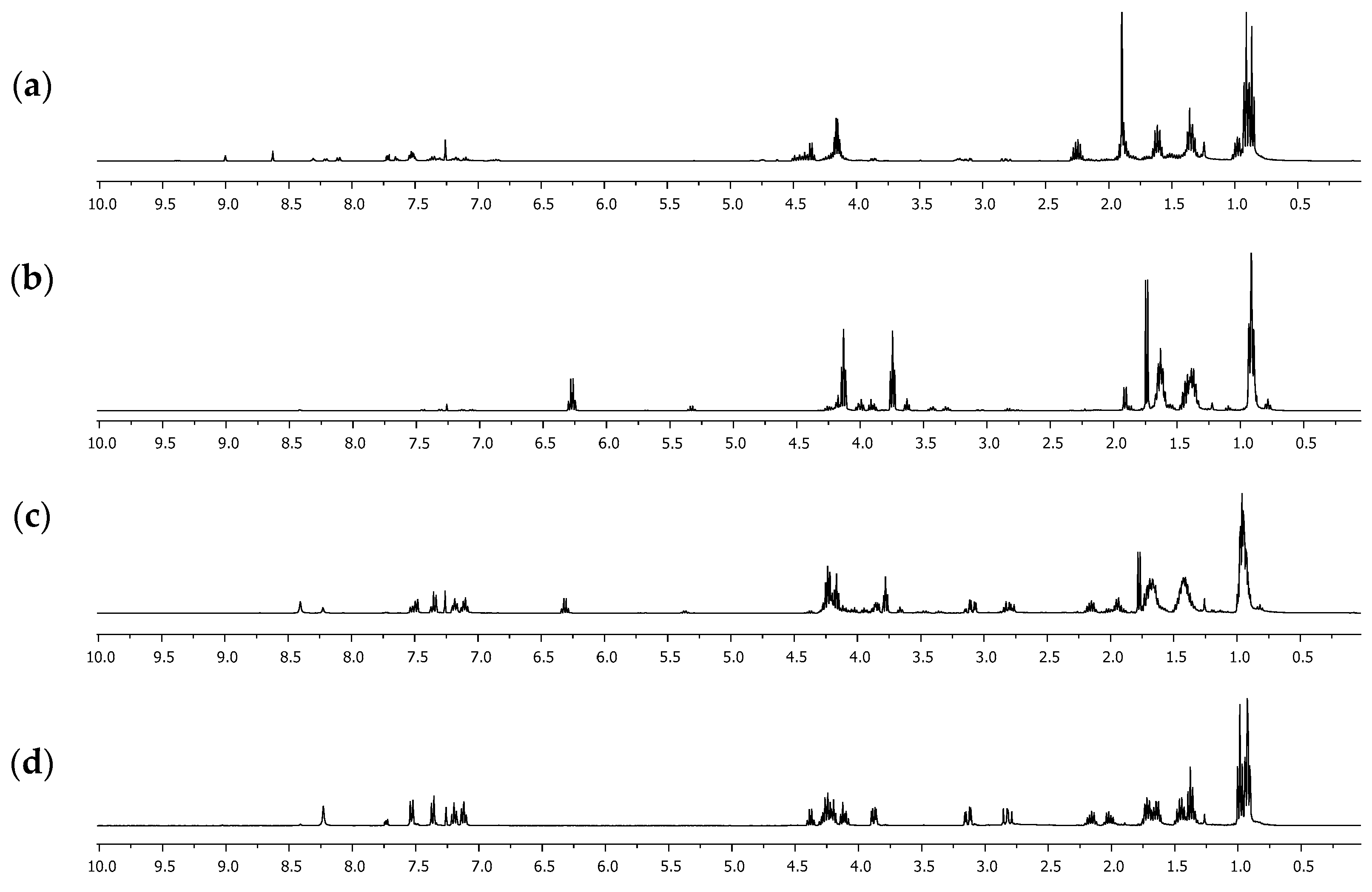
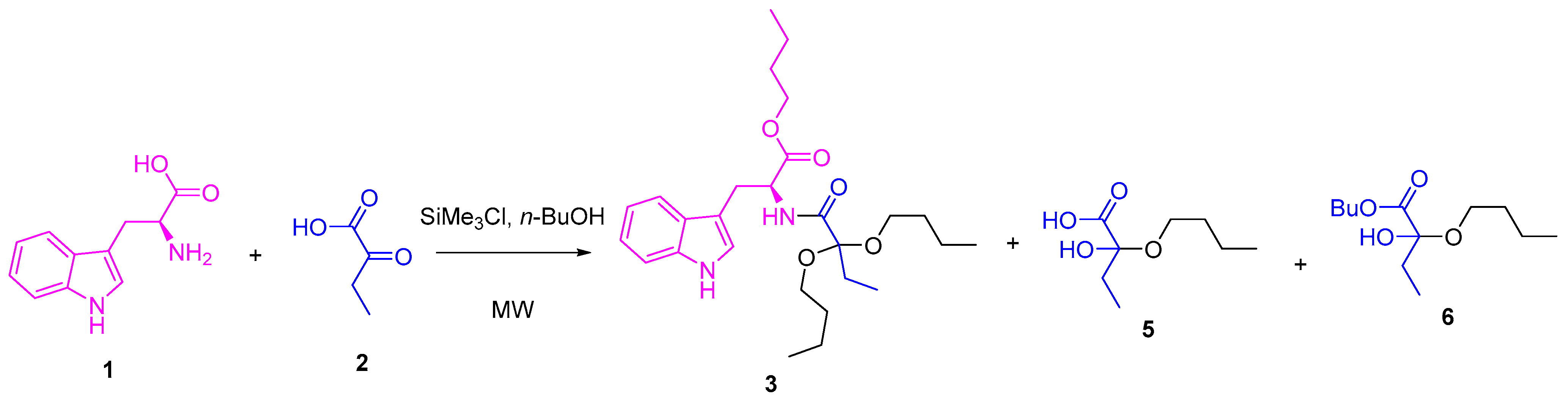
A one-pot methodology was evaluated to improve the yield for compound 3. Thus, mixtures of ʟ-tryptophan 1 and 2-oxobutanoic acid 2 (1 eq:1 eq) in the presence of SiMe3Cl (4 eq) and 1-butanol were heated using MW irradiation at different temperatures, varying the time of reaction (Scheme 2). Other products were detected by 1H NMR of the reaction crudes (Figure 1), which were extracted in each experiment using dichloromethane. The yield of the products depended on the used conditions (Table 2).
Initially, the methodology reported by Quiroga [23], in which alkyl esters derived from ʟ-tryptophan are used as a precursor, was evaluated. The formation of acetals and hemiacetals is determined by the acid-base character of the medium but mainly by the nucleophilic nature of the alcohol used [27]. Bulky alcohols such as 1-butanol have a lower “nucleophilicity” than low-molecular-weight alcohols such as methanol or ethanol. Studies have shown it is possible to use a basic medium that leads to the formation of the corresponding alkoxide [28], which has greater nucleophilicity. A base reagent such as sodium hydroxide was used instead of triethylamine under the same conditions. However, the formation of ʟ-tryptophan 1 as a main product and the presence of the methyl 2-ketobutyrate salt were detected. Since the basic medium does not favor the formation of compound 3, it was decided to modify the synthesis protocol. Instead of using the butyl ester of ʟ-tryptophan 1, the amino acid was used directly as a precursor under a one-pot methodology. Several reports [29,30,31] have shown that one-pot synthesis is a practical methodology and a promising and environmentally friendly approach to modern organic synthesis. Thus, a mixture of ʟ-tryptophan 1 and 2-oxobutanoic acid 2 in 1-butanol reacted in excess of trimethylsilane chloride (SiMe3Cl), a dehydrating agent used in this type of reaction, specifically using amino acids as precursors [32]. Microwave irradiation (MW) was carried out at different temperatures and reaction times, obtaining that the conditions evaluated affected the progress of the reaction, as can be seen in the results of Table 2 and Figure 1. When the temperature considered was 80 °C, the formation of compound 5 was detected, which was identified as a compound with a hemiacetal functional group and carboxylic acid derived exclusively from 2-oxobutanoic acid 2.
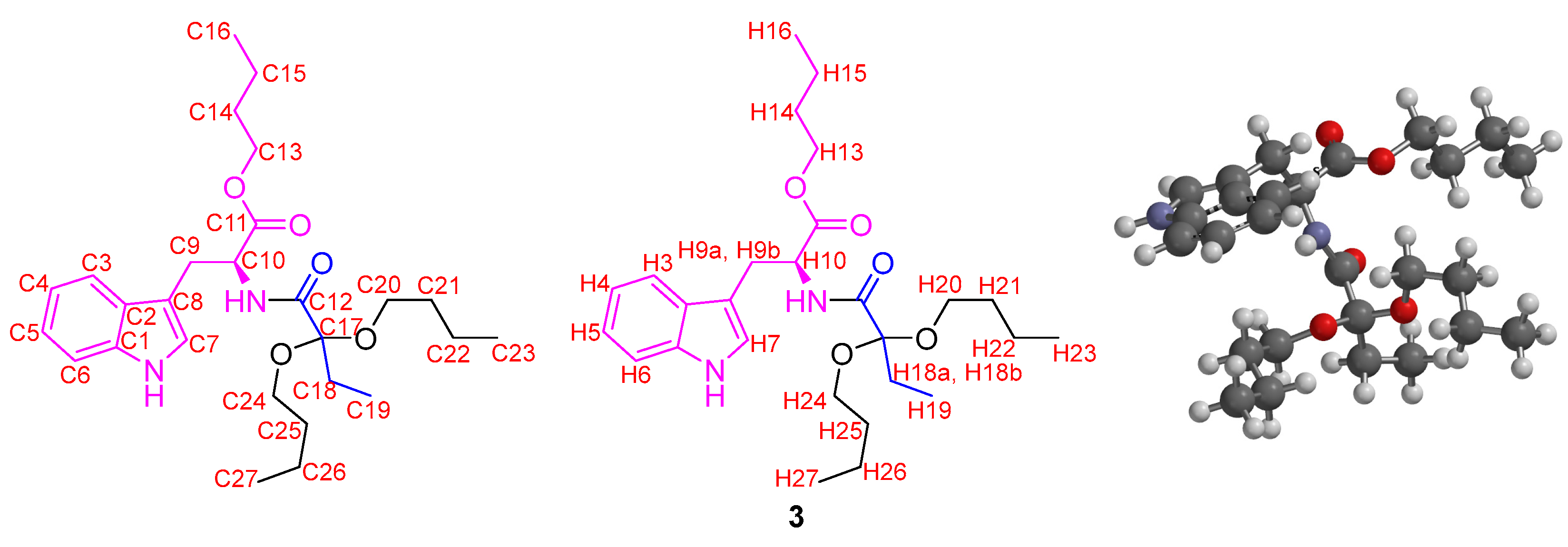
The 1H NMR spectra showed six signals between 0.5 and 5.0 ppm. Between 0.8 and 1.0 ppm, two signals appeared as triplets, with a coupling constant of 7.4 Hz, that integrate for six hydrogen atoms, which correspond to two methyl groups of the ethyl fragment attached to the hemiacetal carbon and the butyl group. The multiplet signals between 1.2 and 1.7 ppm that integrate for two hydrogen atoms correspond to the CH2CH2 methylene groups linked to the methyl group in the butyl fragment. Two multiplet signals were evident between 1.8 and 2.4 ppm, which integrated for one hydrogen atom each, which were assigned to the methylene group of the ethyl group attached to the hemiacetalic carbon and appeared as a pair of signals given that they are diastereotopic hydrogens. Finally, between 4.1 and 4.3 ppm, a signal with triplet multiplicity and a coupling constant of 6.6 Hz, which was assigned to the CH2-O group, was evident. Furthermore, it is possible to affirm that compound 5 is formed as a mixture of the enantiomers since the value of the specific rotation measured was close to 0°. The yield of 5 increases significantly if the reaction time is increased. However, if the reaction temperature rises to 120 °C, the yield of 5 decreases significantly, with product 6 being formed, which was identified as an ester-type derivative with the presence of the hemiacetal. The 1H NMR spectrum showed three overlapping triplet signals around 0.9 ppm, which integrated for nine hydrogen atoms and presented a coupling constant of 7.4 Hz. These signals indicated the presence of three methyl groups, one more than what was observed for compound 5, which was assigned to the methyl group of the additional butyl fragment in the formed acetal functional group. Similarly, multiplet signals between 1.3 and 1.7 ppm increased their relative integral, being assigned for five hydrogen atoms. These signals were also given to the methylene CH2CH2 groups linked to the methyl group of each butyl fragment but also to the diastereotopic hydrogen atoms of the CH2 group in the ethyl fragment attached directly to the hemiacetalic carbon, which, concerning 5, undergo a high-field shift due to an inductive effect. Finally, two triplet signals with a coupling constant of 6.6 Hz and an integral corresponding to two hydrogen atoms were assigned to the methylene CH2-O groups. This result suggested that 6 is formed from 5, through the esterification reaction, which is favored over the hemiacetal formation, presumably by kinetic factors. When the reactions are carried out at a temperature greater than 140 °C, a decrease in the percentage yield of 6 and the appearance of compound 3 as the main product is again evident. This result suggested that 3 is formed from 6 and the ʟ-tryptophan butyl ester 4, which is formed under these conditions but was not detected in the reaction crudes due to its low solubility in dichloromethane. After 40 min of microwave irradiation (MW) at 140 °C, an amidation reaction between 4 and 6 occurred, forming product 3. The 1H NMR spectrum of 3 showed the characteristic signals for the hydrogen atoms of the indole heteroaromatic fragment, which appeared between 7.1 and 8.3 ppm and integrated five hydrogen atoms. Furthermore, the typical signals of benzylic hydrogens presented in ʟ-tryptophan derivatives were evident, which appeared as double doublets between 2.5 and 3.3 ppm, as they were diastereotopic. The hydrogen atom attached to the chiral center appeared as a multiplet at 3.88 ppm. The signals between 4.0–4.4 ppm and 0.8–2.2 ppm were relevant since they demonstrated the formation of the acetal group. Remarkably, the methylene-type hydrogen atoms attached to the “acetalic” carbon atom appeared as two different signals between 2.0 and 2.2 ppm, demonstrating that they were also diastereotopic. Moreover, the 13C NMR spectrum showed acetal carbon as a signal in 110.6 ppm, displaced concerning the remaining aliphatic signals. Fully NMR signals are presented in Figure 2 and Table 3.
The NMR experiment of compound 3 was striking since the signal corresponding to the methylene-type hydrogens linked directly to the “acetalic” carbon appeared as two different signals, demonstrating their diastereotopic character. DFT B3LYP computational calculations at the 6-311G* level were carried out. The calculated and optimized molecular structure of compound 3 showed a total energy value of −1540 a.u., with structural characteristics that determine the magnetic differences between the different carbon and hydrogen atoms. The bond angles around the acetal carbon were 107° for O-C-O and 112° for the C-C-C fragment. The dihedral angle between the C24-O-C17 and C20-O-C17 planes was 167°, showing an antiperiplanar conformation between the butyl groups of the acetal. The dihedral angles between the planes formed through the C=O group of the amide group and each C-O bond of the acetal were 90.5 and 152.0°. These results demonstrated that the adopted structure of 3 presents asymmetric elements, especially in the location of the acetal group. Concerning the NMR calculations, the calculated values of the chemical shifts for the H atoms (Table 3) present minor differences from the experimental values, which are below 1 ppm in most cases.
For the H7 and H25 atoms, it is evident that the difference is around 1 ppm. The H7 atom moved to the low field, while the H25 atom appeared at the high field. The differences found can be explained based on magnetic anisotropy and deprotection due to the inductive effect. For the H7 atom, the inductive effect exerted by the hydrogen of the amino group of the indole ring is presumably marked, which is evident as an exchangeable proton, given that its signal in the 1H NMR spectrum is not evident or appears enormously broadened. Thus, the electronic density of the nitrogen atom decreases, possibly due to intermolecular hydrogen bond formation, making the electronic density at the location of the H7 atom lower and causing an increase in the d of this signal. Regarding the H25 atom, magnetic anisotropy is evident such that the local field currents formed from the carbonyl groups protect these fragments, leading to a lower chemical shift. The calculated molecular structure showed a rigidity that leads to an asymmetry around the acetal carbon, so the hydrogen atoms in the methylenes adjacent to this carbon appeared as different and markedly diastereotopic signals. The presence of multiplets instead of multiplied signals, such as well-defined triplets or quartets, supports this hypothesis. Furthermore, the performed calculations showed that the molecular structure of 3 has a point group C1, which is interpreted in terms of symmetry as a model that does not exhibit any rotational symmetry. This molecule does not have an axis of rotation that leaves its structure invariant, indicating the absence of any axis of symmetry, so there is no axis around which the molecule can rotate and make it coincide with itself.
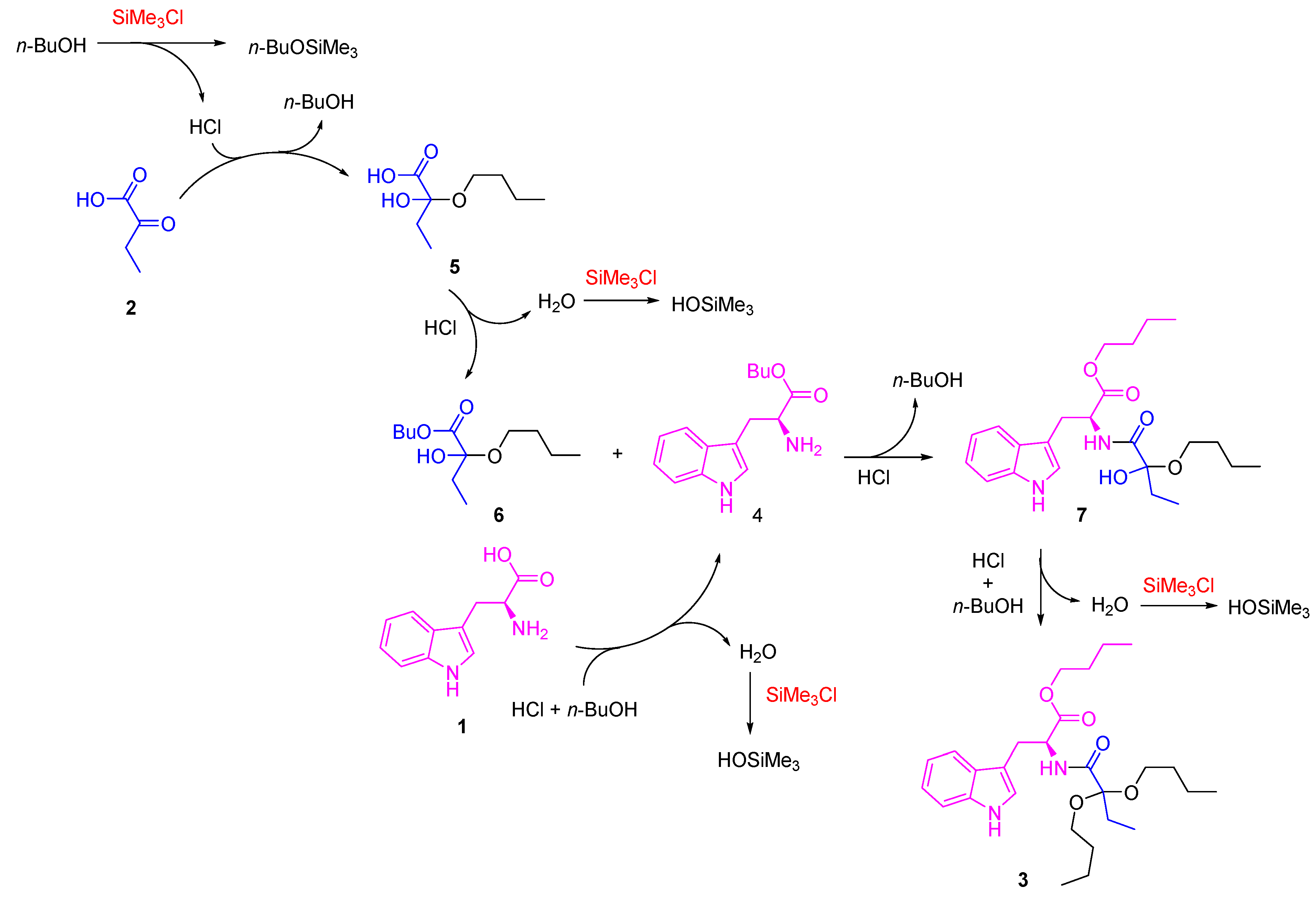
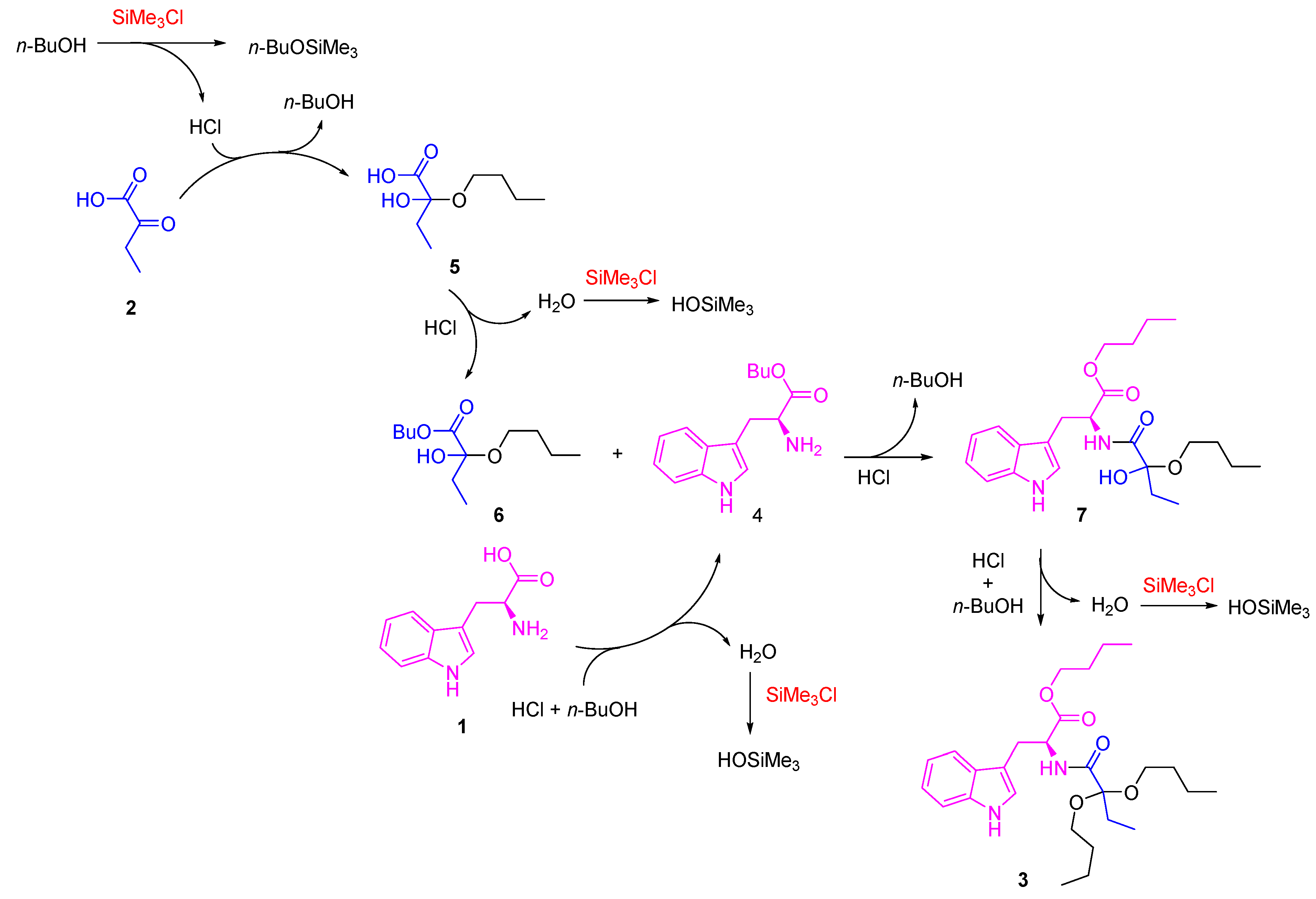
According to these results, a reaction mechanism that follows a series of sequential steps was proposed (Scheme 3). First, the formation of hemiacetal 5 is carried out, which is kinetically favored, as it is a poorly hindered 2-ketocarboxylic acid-type precursor with a high electrophilic character in the ketonic carbon. The presence of SiMe3Cl tends to cause the loss of a water molecule. However, as 1-butanol is not very nucleophilic, the acetal formation is not initially favored. In the second step, the esterification reaction of both ʟ-tryptophan 1 and intermediate 5 occurs, forming compounds 4 and 6, respectively. Subsequently, intermediates 4 and 6 react through an amidation reaction, forming an intermediate 7, which finally reacts with 1-butanol, towards the formation of compound 3. The presence of SiMe3Cl is decisive in the reaction since it favors the elimination of water from the medium in crucial steps [33,34], presumably participating in the final formation of 3 since, as proposed by Izumi, it first reacts with alcohols or carboxylic acids to form HCl, which is probably the definitive catalyst for these reaction systems, in addition to trapping water and thus selectively promoting these dehydration reactions when used in excess.
3. Materials and Methods
3.1. General
All reagents and chemicals were commercially acquired (Merck KGaA and/or Sigma-Aldrich, Darmstadt, Germany). They were employed without additional refinement. As a result, the purity of dry solvents was sufficiently defined during purchase. The products’ progression of reaction and purification were monitored by thin-layer chromatography (TLC) on silica gel 60 F254 plates (Merck KGaA) under detection at 254 nm. Nuclear magnetic resonance (NMR) experiments were conducted using a Bruker Avance AV-400 MHz spectrometer. TMS was used as a reference to give chemical shifts in δ (ppm). Typical splitting patterns were implemented to define the signal multiplicity (i.e., s, singlet; d, doublet; t, triplet; m, multiplet). The chemical reactions were carried out in a Single-mode Discover System microwave reactor model 908,005 series DY1030 in a closed vessel controlling the temperature. Mass spectrometry was carried out on a UHPLC Shimadzu Nexera X2 chromatograph coupled to a QTOF LCMS9030 spectrometer. Formic acid 0.1%: methanol 40:60 was employed as the mobile phase, with a total run time of 20 min, using a Shim-pack HR-ODS column 150 mm L × 3 mm ID, 80 A°. The analysis was performed in positive and negative ion modes, finding that the most appropriate mode was ESI+, which detected the [M+H]+ pseudomolecular ion.
3.2. Chemical Synthesis
A mixture of ʟ-tryptophan (1 mmol) and 2-oxobutanoic acid (1 mmol) in the presence of SiMe3Cl (4 mmol) in excess of 1-butanol (5 mL) was placed in a microwave reactor tube. The mixture was irradiated for 40 min at 140 °C. Then, triethylamine (4.4 mmol) was added, and irradiation was resumed for a further 20 min at the same temperature. The reaction crude was concentrated to dryness by distillation under reduced pressure. Finally, compound 3 was purified by column chromatography, using silica gel as the stationary phase and hexane: ethyl acetate mixtures of variable compositions as the mobile phase. 1H and 13C NMR spectroscopy, ESI-MS, and FT-IR spectroscopy were used to characterize the compound. The results are presented in the Supplementary Materials (Figures S1–S3).
3.3. DFT B3LYP Calculations
Theoretical calculations were performed using the PC Spartan’14 software (Wavefunction Inc., Irvine, CA, USA). The geometric structure of compound 8 was fully optimized without imposing any symmetry constraint with Becke’s three-parameter hybrid functional and the Lee–Yang–Parr correlation functional (B3LYP) at the level 6-311G*.
4. Conclusions
We presented the synthesis of the title compound 3 through a one-pot reaction involving ʟ-tryptophan, 2-oxobutanoic acid, and 1-butanol. Microwave irradiation at varying temperatures and reaction times revealed temperature-sensitive reactions leading to the formation of hemiacetal compound 5 and ester-type derivative 6. The subsequent decrease in the yield of 6 and the appearance of compound 3 at higher temperatures suggested an amidation reaction between intermediates 4 and 6, leading to the formation of the desired product. SiMe3Cl played a crucial role in eliminating water and selectively promoting dehydration reactions, with the proposed involvement in forming HCl as a definitive catalyst in the reaction system.
Supplementary Materials
Spectroscopic data for the characterization of compounds 3, 5, and 6; Figure S1: 1H NMR of compound 3; Figure S2: 13C NMR spectrum of compound 3; Figure S3: ESI+-HRMS spectrum of compound 3; Figure S4: 1H NMR spectrum of compound 5; Figure S5: 13C NMR spectrum of compound 5; Figure S6: 1H NMR spectrum of compound 6.
Author Contributions
Conceptualization, D.Q. and E.C.-B.; methodology, D.Q. and E.C.-B.; software, D.Q. and E.C.-B.; chemical synthesis and structural elucidation, D.Q. and E.C.-B.; writing—original draft preparation, D.Q. and E.C.-B.; writing—review and editing, D.Q. and E.C.-B.; project administration, D.Q. and E.C.-B.; funding acquisition, D.Q. and E.C.-B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was granted by the Universidad Militar Nueva Granada (UMNG). It is a product derived from the project INV-CIAS-2941 funded by Vicerrectoría de Investigaciones at UMNG—Validity 2019.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors thank UMNG for funding this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhou, D.; Porter, W.R.; Zhang, G.G.Z. Drug Stability and Degradation Studies. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2017; pp. 113–149. ISBN 978-0-12-802447-8. [Google Scholar]
- Grindley, T.B.; Gulasekharam, V. Benzylidene Acetal Structural Elucidation by N.M.R. Spectroscopy: Application of Carbon-13. N.M.R.-Spectral Parameters. Carbohydr. Res. 1979, 74, 7–30. [Google Scholar] [CrossRef]
- Orliac, A.; Gomez Pardo, D.; Bombrun, A.; Cossy, J. XtalFluor-E, an Efficient Coupling Reagent for Amidation of Carboxylic Acids. Org. Lett. 2013, 15, 902–905. [Google Scholar] [CrossRef]
- Jereb, M.; Vražič, D.; Zupan, M. Iodine-Catalyzed Transformation of Molecules Containing Oxygen Functional Groups. Tetrahedron 2011, 67, 1355–1387. [Google Scholar] [CrossRef]
- Huang, J.; Lu, Y.; Qiu, B.; Liang, Y.; Li, N.; Dong, D. One-Pot Synthesis of Substituted Isothiazol-3(2 H)-Ones: Intramolecular Annulation of α-Carbamoyl Ketene-S,S-Acetals via PIFA-Mediated N-S Bond Formation. Synthesis 2007, 2007, 2791–2796. [Google Scholar] [CrossRef]
- Sheng, G.; Zhang, W. New Advances of the Methods of Amide Function Group for Construction. Chin. J. Org. Chem. 2013, 33, 2271. [Google Scholar] [CrossRef]
- Masse, C.E.; Yang, M.; Solomon, J.; Panek, J.S. Total Synthesis of (+)-Mycotrienol and (+)-Mycotrienin I: Application of Asymmetric Crotylsilane Bond Constructions. J. Am. Chem. Soc. 1998, 120, 4123–4134. [Google Scholar] [CrossRef]
- Kidjemet, D. N,N-Dimethylformamide Dimethyl Acetal. Synlett 2002, 2002, 1741–1742. [Google Scholar] [CrossRef]
- Kinderman, S.S.; de Gelder, R.; van Maarseveen, J.H.; Schoemaker, H.E.; Hiemstra, H.; Rutjes, F.P.J.T. Amidopalladation of Alkoxyallenes Applied in the Synthesis of an Enantiopure 1-Ethylquinolizidine Frog Alkaloid. J. Am. Chem. Soc. 2004, 126, 4100–4101. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A. Cerium(IV) Ammonium Nitrate: A Versatile Oxidant in Synthetic Organic Chemistry. Synlett 2005, 2005, 3014–3015. [Google Scholar] [CrossRef]
- Lippur, K.; Kanger, T.; Kriis, K.; Kailas, T.; Müürisepp, A.-M.; Pehk, T.; Lopp, M. Synthesis of (2S,2′S)-Bimorpholine N,N′-Quaternary Salts as Chiral Phase Transfer Catalysts. Tetrahedron Asymmetry 2007, 18, 137–141. [Google Scholar] [CrossRef]
- Juma, B.; Adeel, M.; Villinger, A.; Langer, P. Efficient Synthesis of 2,6-Dioxo-1,2,3,4,5,6-Hexahydroindoles Based on the Synthesis and Reactions of (2,4-Dioxocyclohex-1-Yl)Acetic Acid Derivatives. Tetrahedron Lett. 2008, 49, 2272–2274. [Google Scholar] [CrossRef]
- Carmali, S.; Brocchini, S. Polyacetals. In Natural and Synthetic Biomedical Polymers; Elsevier: Amsterdam, The Netherlands, 2014; pp. 219–233. ISBN 978-0-12-396983-5. [Google Scholar]
- Yanev, P.; Angelov, P. Synthesis of Functionalised β-Keto Amides by Aminoacylation/Domino Fragmentation of β-Enamino Amides. Beilstein J. Org. Chem. 2018, 14, 2602–2606. [Google Scholar] [CrossRef] [PubMed]
- Zaher, S.; Christ, L.; Abd El Rahim, M.; Kanj, A.; Karamé, I. Green Acetalization of Glycerol and Carbonyl Catalyzed by FeCl3·6H2O. Mol. Catal. 2017, 438, 204–213. [Google Scholar] [CrossRef]
- Halli, J.; Hofman, K.; Beisel, T.; Manolikakes, G. Synthesis of N-Acyl-N,O-acetals from Aldehydes, Amides and Alcohols. Eur. J. Org. Chem. 2015, 2015, 4624–4627. [Google Scholar] [CrossRef]
- Sattenapally, N.; Sharma, J.; Hou, Y. Selective Conversion of Primary Amides to Esters Promoted by KHSO4. Arkivoc 2018, 2018, 174–183. [Google Scholar] [CrossRef]
- Miranda, L.P.; Jones, A.; Meutermans, W.D.F.; Alewood, P.F. P-Cresol As a Reversible Acylium Ion Scavenger in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1998, 120, 1410–1420. [Google Scholar] [CrossRef]
- Vincent, S.; Lebeau, L.; Mioskowski, C. N, N-Dibenzyl Formamide Dimethyl Acetal and N, N-Dibenzyl Chloromethylene Iminium Chloride: Two Complementary Reagents for the Protection Of Primary Amines as N, N-Dibenzyl Formamidines. Synth. Commun. 1999, 29, 167–174. [Google Scholar] [CrossRef]
- Tsotinis, A.; Vlachou, M.; Kiakos, K.; Hartley, J.A.; Thurston, D.E. Design and Synthesis of Two Cytotoxic Analogs of the Novel Pyrrolo[1′,2′:1,2][1,4]Diazepin [7,6-b]Indol-5(6H)-One Nucleus. Chem. Lett. 2003, 32, 512–513. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Li, H.-J.; Liu, L.; Demoulin, N.; Liu, Z.; Wang, D.; Chen, Y.-J. Facile Synthesis of Spiropyrans from Chromene Hemiacetal Esters and Bifunctional Nucleophiles. Synlett 2011, 2011, 1573–1578. [Google Scholar] [CrossRef]
- Luo, C.; Xu, Q.; Huang, C.; Luo, L.; Zhu, J.; Zhang, R.; Huang, G.; Yin, D. Development of an Efficient Synthetic Process for Broflanilide. Org. Process Res. Dev. 2020, 24, 1024–1031. [Google Scholar] [CrossRef]
- Quiroga, D. Employing Molecular Docking Calculations for the Design of Alkyl (2-Alcoxy-2-Hydroxypropanoyl)-ʟ-Tryptophanate Derivatives as Potential Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1). Reactions 2023, 4, 108–116. [Google Scholar] [CrossRef]
- Ma, X.-Y.; Shao, F.-Q.; Hu, X.; Liu, X. Progress in the Synthesis of N-Acyl-N,O-Acetals. Synthesis 2022, 54, 1203–1216. [Google Scholar] [CrossRef]
- Iwai, K.; Hikasa, A.; Yoshioka, K.; Tani, S.; Umezu, K.; Nishiwaki, N. Synthesis of Bis(Functionalized) Aminals via Successive Nucleophilic Amidation and Amination. J. Org. Chem. 2023, 88, 2207–2213. [Google Scholar] [CrossRef]
- Quiroga, D.; Becerra, L.; Sadat-Bernal, J.; Vargas, N.; Coy-Barrera, E. Synthesis and Antifungal Activity against Fusarium Oxysporum of Some Brassinin Analogs Derived from ʟ-Tryptophan: A DFT/B3LYP Study on the Reaction Mechanism. Molecules 2016, 21, 1349. [Google Scholar] [CrossRef]
- Liu, B.; Thayumanavan, S. Substituent Effects on the pH Sensitivity of Acetals and Ketals and Their Correlation with Encapsulation Stability in Polymeric Nanogels. J. Am. Chem. Soc. 2017, 139, 2306–2317. [Google Scholar] [CrossRef]
- Phan, T.B.; Mayr, H. Comparison of the Nucleophilicities of Alcohols and Alkoxides. Can. J. Chem. 2005, 83, 1554–1560. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, F.-E. One-Pot Synthesis and Its Practical Application in Pharmaceutical Industry. Curr. Org. Synth. 2012, 9, 873–897. [Google Scholar] [CrossRef]
- Hayashi, Y. Pot Economy and One-Pot Synthesis. Chem. Sci. 2016, 7, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, W. Recent Developments in One-Pot Stepwise Synthesis (OPSS) of Small Molecules. iScience 2022, 25, 105005. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sha, Y. A Convenient Synthesis of Amino Acid Methyl Esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Fukase, K.; Kusumoto, S. TMSCl as a Mild and Effective Source of Acidic Catalysis in Fischer Glycosidation and Use of Propargyl Glycoside for Anomeric Protection. Biosci. Biotechnol. Biochem. 2002, 66, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.J.; Saccomando, D.J.; Walker, G.; Mansell, S.M. Sustainable Routes to Alkenes: Applications of Homogeneous Catalysis to the Dehydration of Alcohols to Alkenes. Catal. Sci. Technol. 2023, 13, 2638–2647. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of compound 3 using the previously reported methodology [23].
Scheme 1.
Synthesis of compound 3 using the previously reported methodology [23].
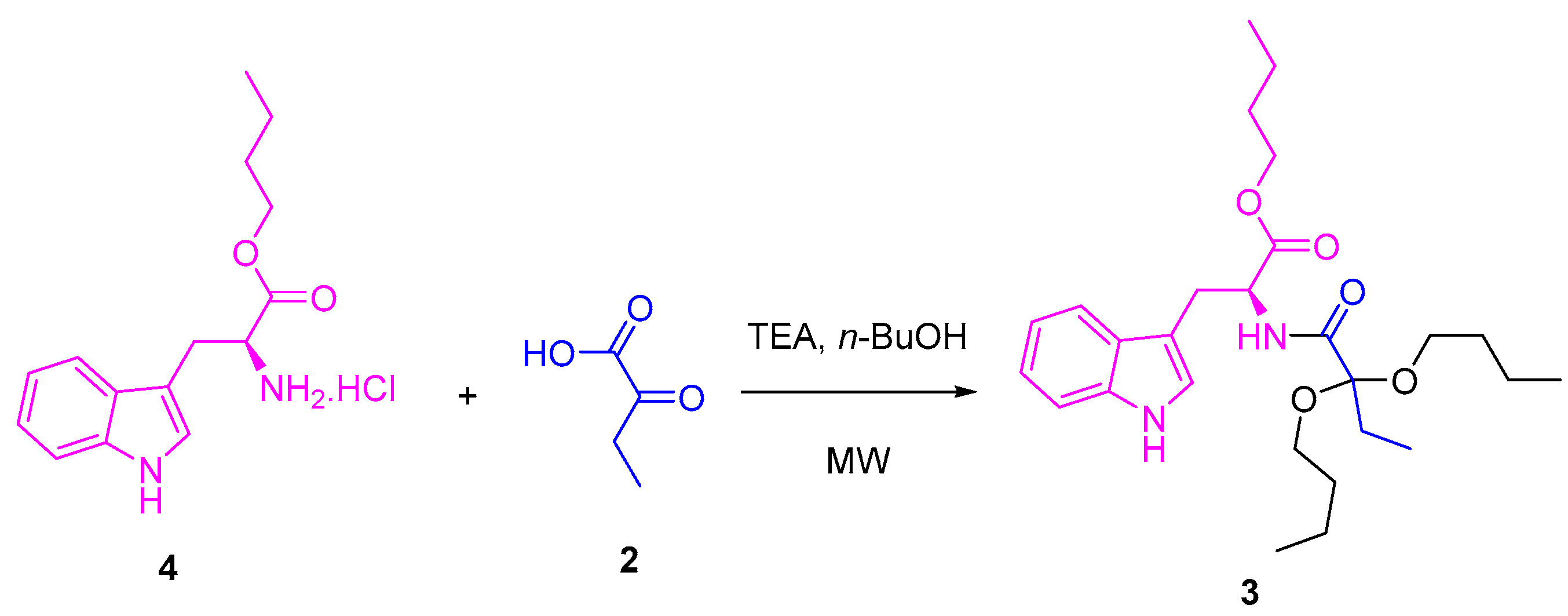
Scheme 2.
One-pot synthesis of compound 3.

Figure 1.
1H NMR spectra of the extracted reaction crudes at different temperatures in CDCl3: (a) 80 °C, 40 min; (b) 120 °C, 40 min; (c) 140 °C, 20 min; (d) 140 °C, 40 min.
Figure 1.
1H NMR spectra of the extracted reaction crudes at different temperatures in CDCl3: (a) 80 °C, 40 min; (b) 120 °C, 40 min; (c) 140 °C, 20 min; (d) 140 °C, 40 min.
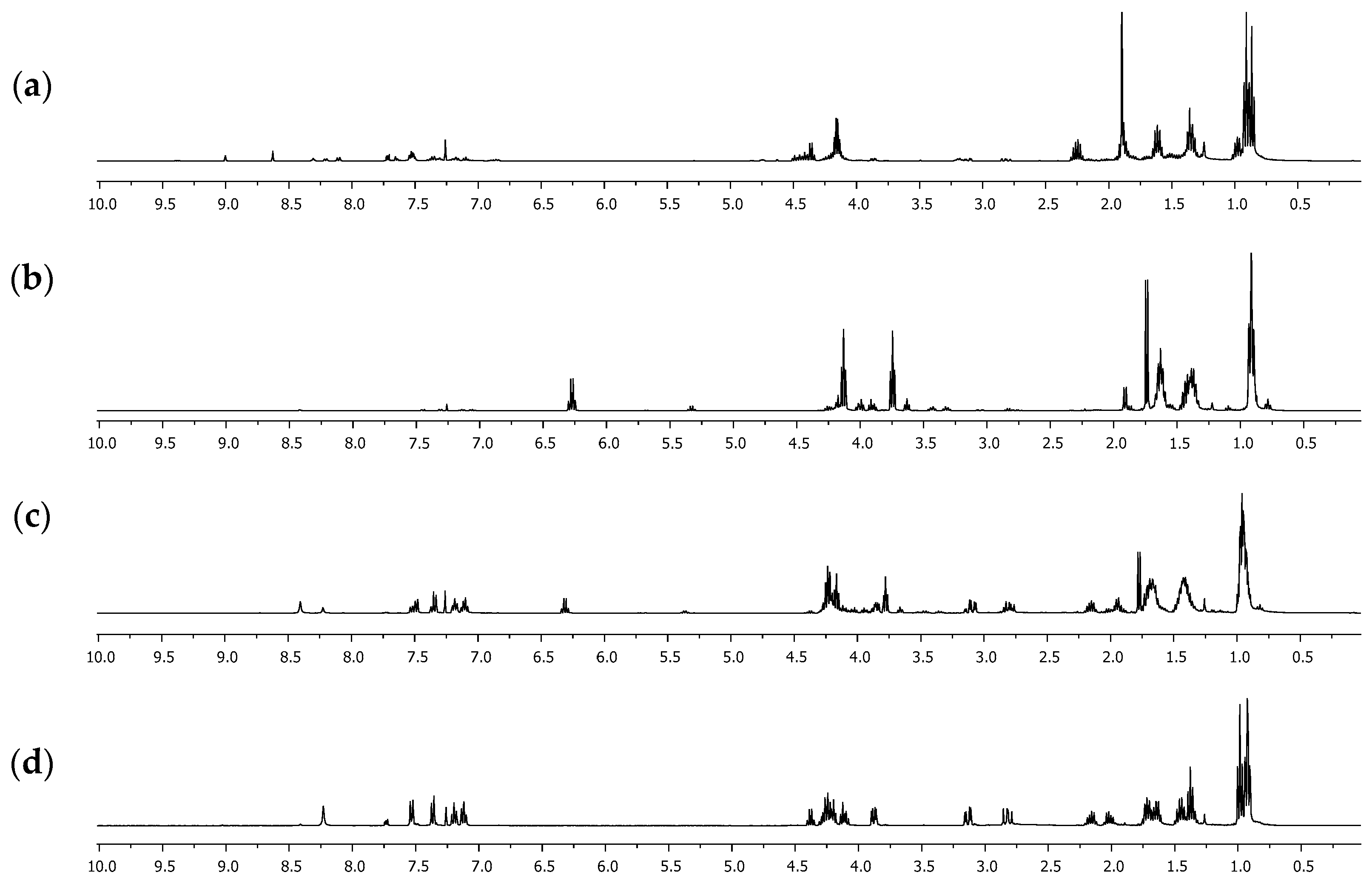
Figure 2.
Labeled chemical structure and calculated optimized molecular structure of 3.

Scheme 3.
Proposed reaction mechanism for the formation of compound 3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
MW-assisted synthesis of compound 3 from four varying reaction conditions.
| Entry | Conditions | Yield of 3 (%) |
|---|---|---|
| 1 | 80 °C, 20 min | No detected |
| 2 | 80 °C, 40 min | No detected |
| 3 | 120 °C, 20 min | 5 |
| 4 | 120 °C, 40 min | 7 |
| 5 | 140 °C, 20 min | 13 |
| 6 | 140 °C, 40 min | 22 |
Table 2.
One-pot MW-assisted synthesis of compound 3 varying reaction conditions.
| Entry | Conditions | Yield of 3 (%) | Yield of 5 (%) | Yield of 6 (%) |
|---|---|---|---|---|
| 1 | 80 °C, 20 min | No detected | 15 | 2 |
| 2 | 80 °C, 40 min | No detected | 31 | 4 |
| 3 | 120 °C, 20 min | No detected | 3 | 23 |
| 4 | 120 °C, 40 min | No detected | No detected | 51 |
| 5 | 140 °C, 20 min | 47 | No detected | 25 |
| 6 | 140 °C, 40 min | 89 | No detected | No detected |
Table 3.
Experimental and calculated chemical shifts of the carbon and hydrogen atoms of compound 3.
Table 3.
Experimental and calculated chemical shifts of the carbon and hydrogen atoms of compound 3.
| C | Experimental δC | Calculated δC | ΔδC | H | Experimental δH | Calculated δH | ΔδH |
|---|---|---|---|---|---|---|---|
| C1 | 136.7 | 141.3 | 4.6 | H3 | 7.53 | 7.69 | 0.16 |
| C2 | 132.0 | 134.5 | 2.4 | H4 | 7.37 | 7.37 | 0.00 |
| C3 | 122.35 | 121.9 | 0.5 | H5 | 7.13 | 7.39 | 0.26 |
| C4 | 126.6 | 125.2 | 1.4 | H6 | 7.20 | 7.31 | 0.11 |
| C5 | 129.0 | 127.3 | 1.7 | H7 | 8.23 | 7.00 | 1.23 |
| C6 | 118.6 | 114.3 | 4.3 | H9a | 2.84 | 3.11 | 0.27 |
| C7 | 119.7 | 117.6 | 2.1 | H9b | 3.14 | 3.17 | 0.03 |
| C8 | 131.0 | 126.4 | 4.6 | H10 | 3.88 | 3.32 | 0.56 |
| C9 | 30.7 | 30.0 | 0.7 | H13 | 4.20 | 3.62 | 0.58 |
| C10 | 54.3 | 63.9 | 9.6 | H14 | 1.41 | 2.06 | 0.65 |
| C11 | 174.1 | 177.5 | 3.4 | H15 | 1.41 | 1.23 | 0.18 |
| C12 | 172.9 | 174.2 | 1.3 | H16 | 0.99 | 1.12 | 0.13 |
| C13 | 65.7 | 69.5 | 3.8 | H18a | 2.06 | 1.87 | 0.19 |
| C14 | 30.8 | 34.5 | 3.7 | H18b | 2.01 | 1.37 | 0.64 |
| C15 | 14.2 | 23.9 | 9.7 | H19 | 0.93 | 1.32 | 0.39 |
| C16 | 8.2 | 16.3 | 8.1 | H20 | 4.10 | 4.11 | 0.01 |
| C17 | 110.6 | 106.3 | 4.3 | H21 | 1.41 | 1.97 | 0.56 |
| C18 | 33.7 | 35.4 | 1.7 | H22 | 1.41 | 1.47 | 0.06 |
| C19 | 13.9 | 11.6 | 2.3 | H23 | 0.99 | 1.06 | 0.07 |
| C20 | 63.2 | 65.7 | 2.5 | H24 | 4.27 | 3.74 | 0.53 |
| C21 | 30.7 | 36.9 | 6.2 | H25 | 1.41 | 2.48 | 1.07 |
| C22 | 25.4 | 25.5 | 0.1 | H26 | 1.41 | 1.48 | 0.07 |
| C23 | 19.3 | 16.7 | 2.6 | H27 | 0.99 | 0.85 | 0.14 |
| C24 | 65.2 | 65.8 | 0.6 | ||||
| C25 | 30.7 | 35.4 | 4.7 | ||||
| C26 | 25.4 | 25.1 | 0.3 | ||||
| C27 | 19.3 | 16.0 | 3.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Quiroga, D.; Coy-Barrera, E. Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate. Molbank 2024, 2024, M1794. https://doi.org/10.3390/M1794
AMA Style
Quiroga D, Coy-Barrera E. Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate. Molbank. 2024; 2024(1):M1794. https://doi.org/10.3390/M1794
Chicago/Turabian StyleQuiroga, Diego, and Ericsson Coy-Barrera. 2024. "Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate" Molbank 2024, no. 1: M1794. https://doi.org/10.3390/M1794
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.