
Diisoamyl (1R, 4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate

 and
and
Abstract
:1. Introduction
2. Results and Discussion
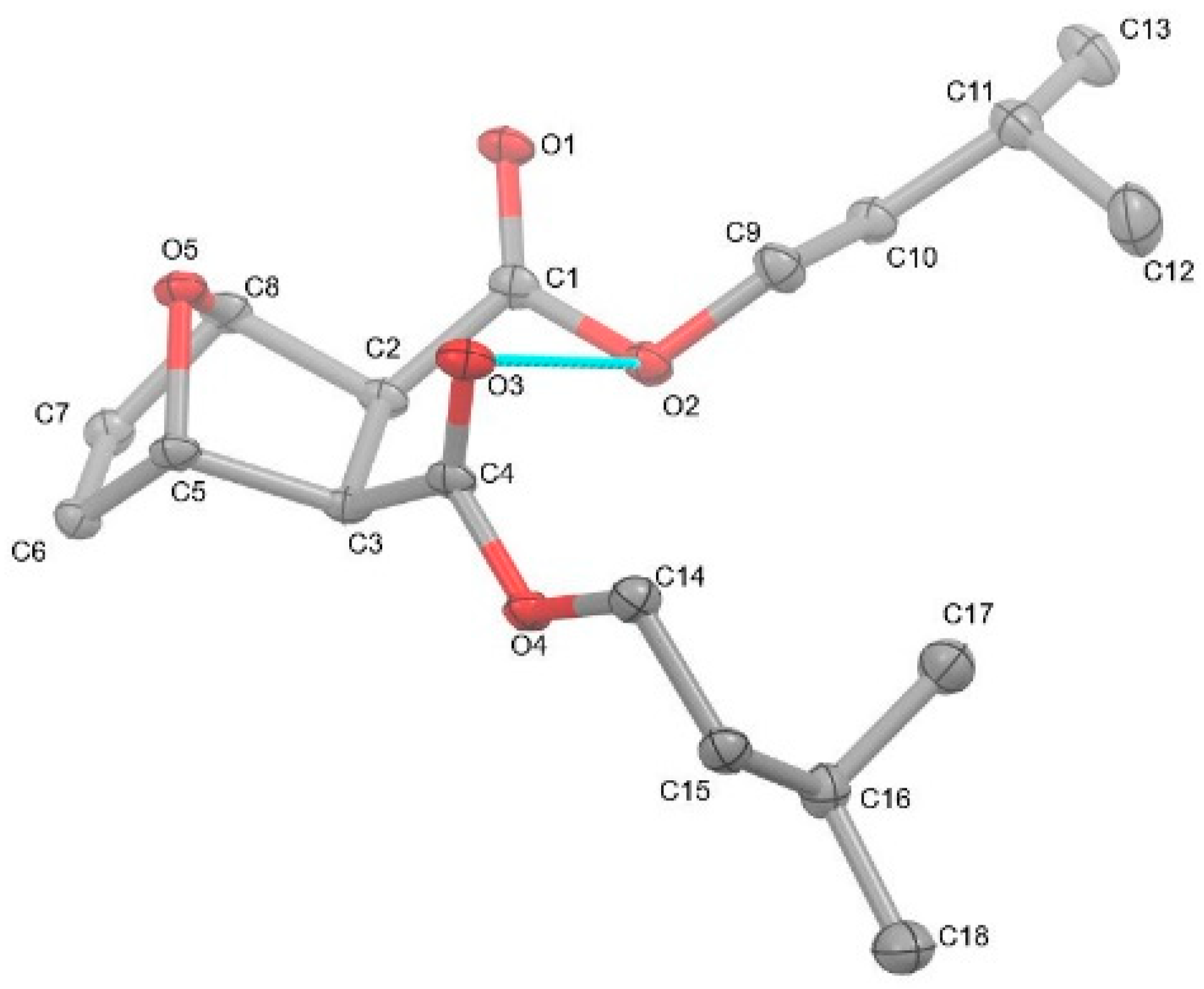
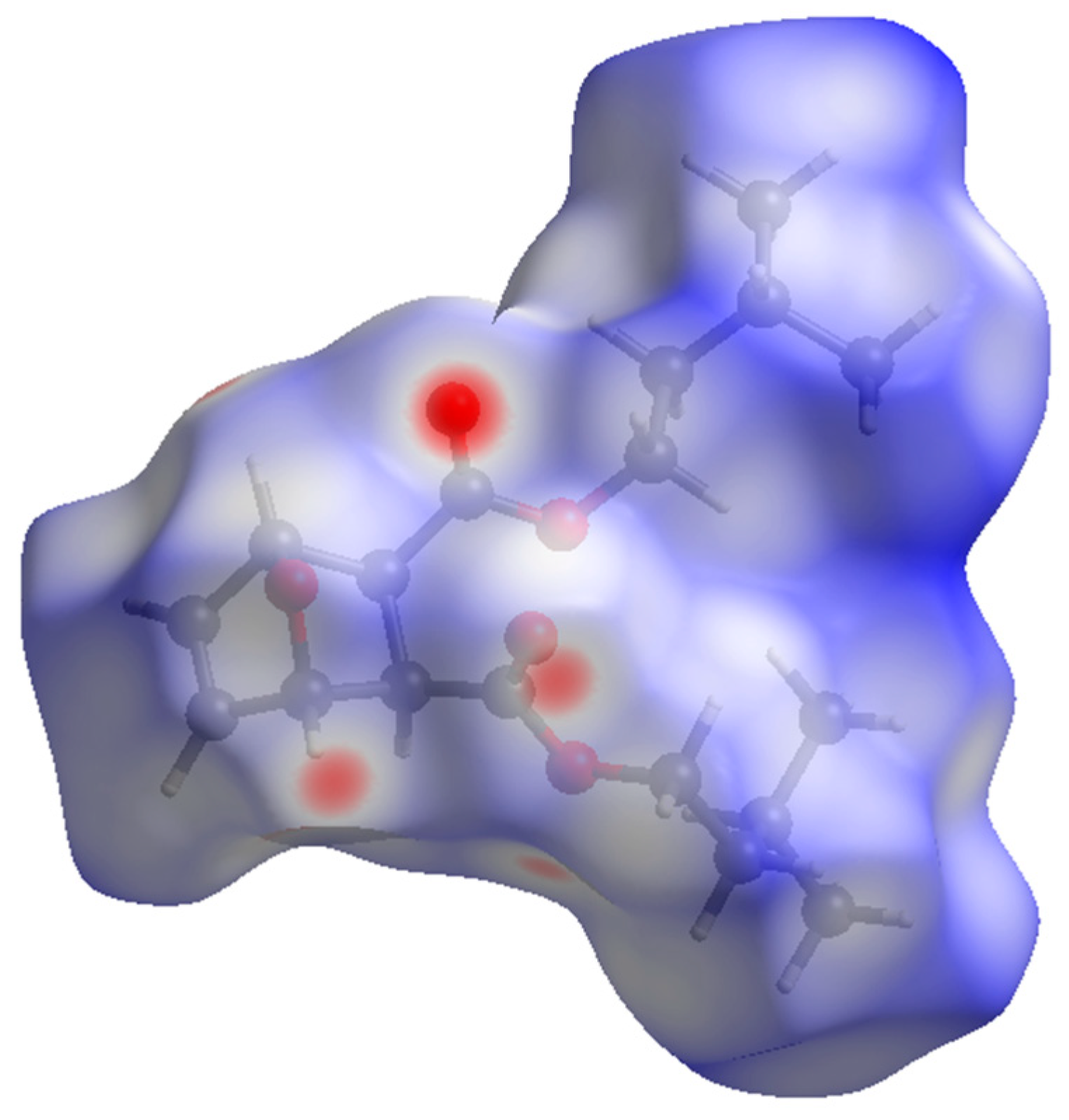
2.1. X-ray Diffraction Studies
2.2. Spectroscopy and Characterization
3. Materials and Methods
3.1. General
3.2. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- McMurry, J. Fundamentals of Organic Chemistry; Brooks/Cole: Monterey, CA, USA, 1986. [Google Scholar]
- Khan, Z.; Javed, F.; Shamair, Z.; Hafeez, A.; Fazal, T.; Aslam, A.; Zimmerman, W.B.; Rehman, F. Current developments in esterification reaction: A review on process and parameters. J. Ind. Eng. Chem. 2021, 103, 80–101. [Google Scholar] [CrossRef]
- Trnka, T.M.; Grubbs, R.H. The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Ulman, M.; Grubbs, R.H. Relative Reaction Rates of Olefin Substrates with Ruthenium(II) Carbene Metathesis Initiators. Organometallics 1998, 17, 2484–2489. [Google Scholar] [CrossRef]
- Grubbs, R.B.; Grubbs, R.H. 50th Anniversary Perspective: Living Polymerization—Emphasizing the Molecule in Macromolecules. Macromolecules 2017, 50, 6979–6997. [Google Scholar] [CrossRef]
- Novak, B.M.; Grubbs, R.H. The Ring-Opening Metathesis Polymerization of 7-Oxabicyclo[2.2.1]Hept-5-Ene Derivatives—A New Acyclic Polymeric Ionophore. J. Am. Chem. Soc. 1988, 110, 960–961. [Google Scholar] [CrossRef]
- Novak, B.M.; Grubbs, R.H. Catalytic Organometallic Chemistry in Water: The Aqueous Ring-Opening Metathesis Polymerization of 7-Oxanorbornene Derivatives. J. Am. Chem. Soc. 1988, 110, 7542–7543. [Google Scholar] [CrossRef]
- Holerca, M.N.; Peterca, M.; Partridge, B.E.; Xiao, Q.; Lligadas, G.; Monteiro, M.J.; Percec, V. Monodisperse Macromolecules by Self-Interrupted Living Polymerization. J. Am. Chem. Soc. 2020, 142, 15265–15270. [Google Scholar] [CrossRef]
- deRonde, B.M.; Posey, N.D.; Otter, R.; Caffrey, L.M.; Minter, L.M.; Tew, G.N. Optimal Hydrophobicity in Ring-Opening Metathesis Polymerization-Based Protein Mimics Required for siRNA Internalization. Biomacromolecules 2016, 17, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Kyu, A.J.; Lim, K.H.; Chul, S.K.; Hoon, L.S. Catalysts for Polymerization or Copolymerization of Olefins, Its preparing Method and Olefins Polymerization or Copolymerization Method. KR20120077690A, 10 July 2012. [Google Scholar]
- Karlen, T.; Ludi, A.; Mühlebach, A.; Bernhard, P.; Pharisa, C. Photoinduced ring opening metathesis polymerization (PROMP) of strained bicyclic olefins with ruthenium complexes of the type [(η6-arene1)Ru(η6-arene2)]2+ and [Ru(Nc-R)6]2+. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 1665–1674. [Google Scholar] [CrossRef]
- Kotsuki, H.; Nishizawa, H.; Ochi, M.; Matsuoka, K. High Pressure Organic Chemistry. V. Diels-Alder Reactions of Furan with Acrylic and Maleic Esters. Bull. Chem. Soc. Jpn. 2006, 55, 496–499. [Google Scholar] [CrossRef]
- Baggio, S.; Barriola, A.; de Perazzo, P.K. Crystal and molecular structure of 7-oxabicyclo[2,2,1]hept-5-ene-2,3-exo-dicarboxylic anhydride. J. Chem.Soc. Perkin Trans. 2 1972, 934–937. [Google Scholar] [CrossRef]
- France, M.B.; Alty, L.T.; Earl, T.M. Synthesis of a 7-oxanorbornene derivative: A two-step sequence preparation for the organic laboratory. J. Chem. Educ. 1999, 76, 659–660. [Google Scholar] [CrossRef]
- Li, J.J. (Ed.) Fischer–Speier esterification. In Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications Fifth Edition; Springer International Publishing: Cham, Switzerland, 2014; p. 252. [Google Scholar]
- Martínez, R.F.; Cravotto, G.; Cintas, P. Organic Sonochemistry: A Chemist’s Timely Perspective on Mechanisms and Reactivity. J. Org. Chem. 2021, 86, 13833–13856. [Google Scholar] [CrossRef]
- Flannigan, D.J.; Suslick, K.S. Inertially confined plasma in an imploding bubble. Nat. Phys. 2010, 6, 598–601. [Google Scholar] [CrossRef]
- Miró Vera, A.; Velásquez, W.; Briceño, A.; Bahsas Bahsas, A.; Ramírez Valero, B.; Diaz de Delgado, G. Synthesis and Crystal Structure of Dimethyl-7-oxabicyclo[2.2.1]hept-5-ene exo,exo-2,3-dicarboxylate. J. Chem. Crystallogr. 2007, 37, 543–548. [Google Scholar] [CrossRef]
- Li, J. Benzothiazol-2-amine-3-methoxycarbonyl-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylic acid (1/1). Acta Crystallogr. Sect. E 2011, 67, o199. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Goh, R.Y.W. CCDC 1027454. CSD Communication 2014. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/cc13h4pf&sid=DataCite (accessed on 11 July 2024). [CrossRef]
- Sadeghi-Khomami, A.; Blake, A.J.; Wilson, C.; Thomas, N.R. Synthesis of a Carbasugar Analogue of a Putative Intermediate in the UDP-Galp-Mutase Catalyzed Isomerization. Org. Lett. 2005, 7, 4891–4894. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Minch, M.J. Orientational dependence of vicinal proton-proton NMR coupling constants: The Karplus relationship. Concepts Magn. Reson. 1994, 6, 41–56. [Google Scholar] [CrossRef]
- McLafferty, F.W. Mass Spectrometric Analysis. Molecular Rearrangements. Anal. Chem. 1959, 31, 82–87. [Google Scholar] [CrossRef]
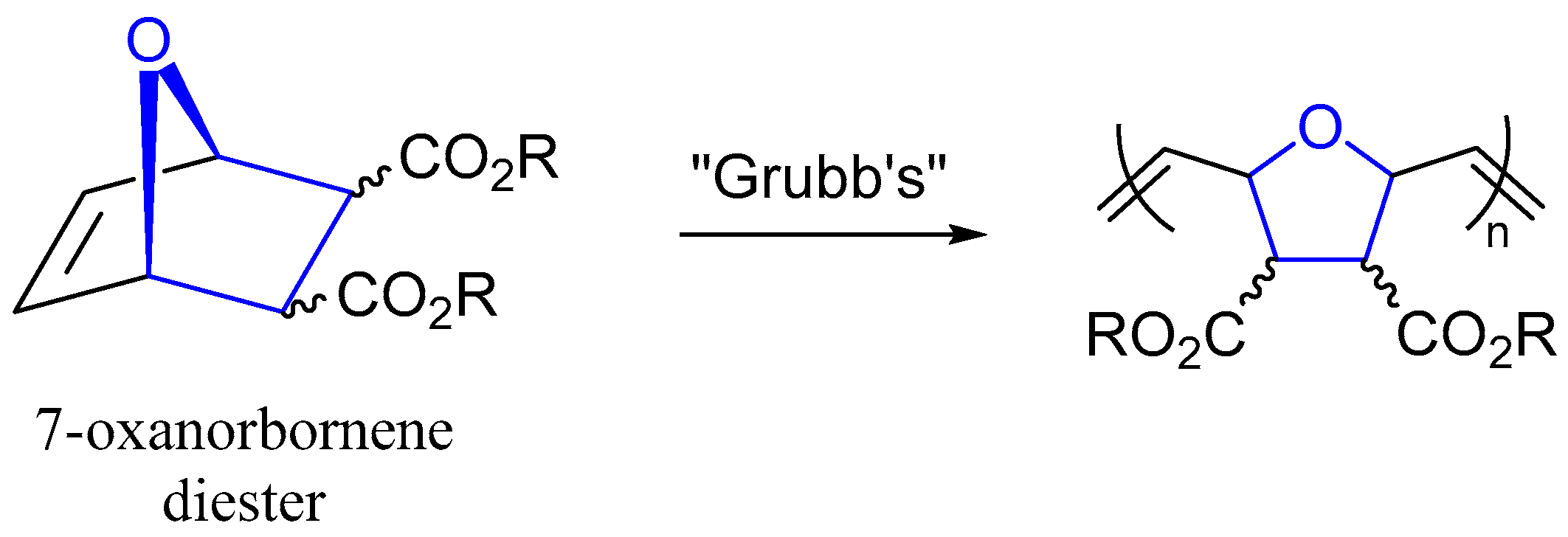



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bonds | Distances | Atoms | Angles |
|---|---|---|---|
| C1–O1 | 1.2076(19) | O1–C1–C2 | 125.90(15) |
| C1–O2 | 1.350(2) | O1–C1–O2 | 123.28(15) |
| C4–O3 | 1.2041(19) | O2–C1–C2 | 110.8(13) |
| C4–O4 | 1.3494(19) | O3–C4–C3 | 125.79(14) |
| O2–C9 | 1.449(2) | O3–C4–O4 | 123.55(15 |
| O4–C14 | 1.4532(19) | O4–C4–C14 | 115.59(12) |
| C6–C7 | 1.329(2) | C5–O5–O8 | 95.95(11) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quillian, B.; Musso, K.; Vinson, E.M.; Bazemore, J.G.; Marks, A.R.; Padgett, C.W. Diisoamyl (1R, 4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate. Molbank 2024, 2024, M1852. https://doi.org/10.3390/M1852
Quillian B, Musso K, Vinson EM, Bazemore JG, Marks AR, Padgett CW. Diisoamyl (1R, 4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate. Molbank. 2024; 2024(3):M1852. https://doi.org/10.3390/M1852
Chicago/Turabian StyleQuillian, Brandon, Kennedy Musso, Elizabeth M. Vinson, Joseph G. Bazemore, Allison R. Marks, and Clifford W. Padgett. 2024. "Diisoamyl (1R, 4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate" Molbank 2024, no. 3: M1852. https://doi.org/10.3390/M1852
APA StyleQuillian, B., Musso, K., Vinson, E. M., Bazemore, J. G., Marks, A. R., & Padgett, C. W. (2024). Diisoamyl (1R, 4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate. Molbank, 2024(3), M1852. https://doi.org/10.3390/M1852