Abstract
Diisoamyl (1R,4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (2) was prepared by reacting exo-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride (1) with isoamyl alcohol in the presence of a sulfuric acid catalyst under sonication conditions. Compound 2 was characterized by 1H, 13C NMR, DEPT-135, infrared, and UV-vis spectroscopy. Gas chromatography–mass spectrometry, elemental analysis, and melting point determination were used to assess purity. The structure of compound 2 was also determined by single-crystal X-ray diffraction. It crystallizes in the monoclinic space group P21/c (14) with cell values of a = 15.5647(3) Å, b = 12.8969(2) Å, c = 9.0873(2) Å; β= 99.3920(10)°.
1. Introduction
The Fisher Esterification reaction is often used to expose sophomore-level organic chemistry students to the nucleophilic acyl substitution reaction [1]. While the reaction may be thought of as esoteric to these students, it is commonly used for the manufacture of pharmaceuticals, biofuels, fragrances, and other important fine- and commodity-grade chemicals on an industrial scale [2]. We became interested in preparing various 7-oxanorbornene diesters to serve as olefin monomers in ring-opening metathesis polymerization (ROMP) reactions using Grubb’s catalysis (Figure 1) [3,4,5]. The impetus of this study was to assess the impact of the ester substituents on living polymerization catalysis rates and polymer monodispersity [3,4,5]. ROMP catalysis using a myriad of 7-oxanorbornene diesters has been shown to produce helical polymers that act as ionophores for dyes [6,7]. Recent advances have shown ROMP catalysis of a sterically encumbered 7-oxanorbornene with dendritic ester R-groups promoted “self-interruption” of the living polymerization to produce polymers of good monodispersity [8]. The literature shows 7-oxanorbornene diesters of great variety (methyl [9], ethyl [8,9], butyl [9,10], benzyl [9], cyclohexyl [9], isobutyl [9,10], phenyl [9], propyl [9,11], hexyl [11]) have been prepared for ROMP catalysis. Surprisingly, the isoamyl diester variant was absent from the literature. Notably, high-pressure reaction techniques have been used to make numerous 7-oxanorbornene diesters [12]. We set out to fill this gap and provide a synthesis and structural determination of diisoamyl (1R,4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (2); the missing link in the repertoire of available 7-oxanorbornene diesters that can be utilized in Grubb’s catalysis.
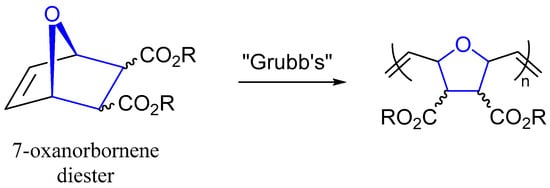
Figure 1.
Generic structure of 7-oxanorbornene diesters and Grubb’s catalysis.
2. Results and Discussion
Compound 2 was prepared from a modified procedure (Figure 2) that called for the reaction of exo-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic (1) with methanol under HCl catalysis and refluxing for two hours [13]. The reaction times were too excessive to accommodate the time allotted for laboratory classes [14]. Indeed, the Fisher Esterification reaction is hampered by its small equilibrium constant (keq = ~4); thus, several strategies have historically been employed to improve the yield of the ester product, which include increased reaction temperatures, use of a catalyst, conducting the reaction in excess alcohol, and the removal of water using a Dean–Stark apparatus or desiccants [15]. Notably, the condensation reaction rate retards with increasing steric bulk of the alcohol or carboxylic acid; therefore, forming compound 2 using this method would prove difficult. To overcome these deficiencies, we adopted sonication techniques [16] to exploit the properties of cavitation chemistry that have been theorized to produce microscopic zones of extreme temperatures and pressures [17]. In concert with sonication, super absorbent polymer beads (sodium polyacrylate beads, Jelly Marbles®) were used as a desiccant to drive the reaction to product formation. To our knowledge, this is the first use of these conditions in the Fisher Esterification reaction. Compound 2 was successfully prepared by reacting isoamyl alcohol with compound 1 and a catalytic amount of sulfuric acid in the presence of gel beads under sonication at 49 °C for 30 min (Figure 2). After workup, compound 2 was isolated as colorless thin crystals in ~20% yield. It should be noted that conducting the reaction under thermal conditions (no sonication) promoted the retro-Diels–Alder reaction to produce diisoamyl maleate and its hydrolyzed product, maleic acid. This decomposition becomes more problematic with temperatures above 50 °C. It should also be noted that methyl, ethyl, and isopropyl analogs of the 7-oxanorbornene diesters can be prepared using these standard conditions.
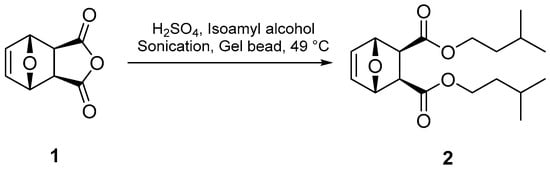
Figure 2.
Line reaction for the formation of compound 2.
2.1. X-ray Diffraction Studies
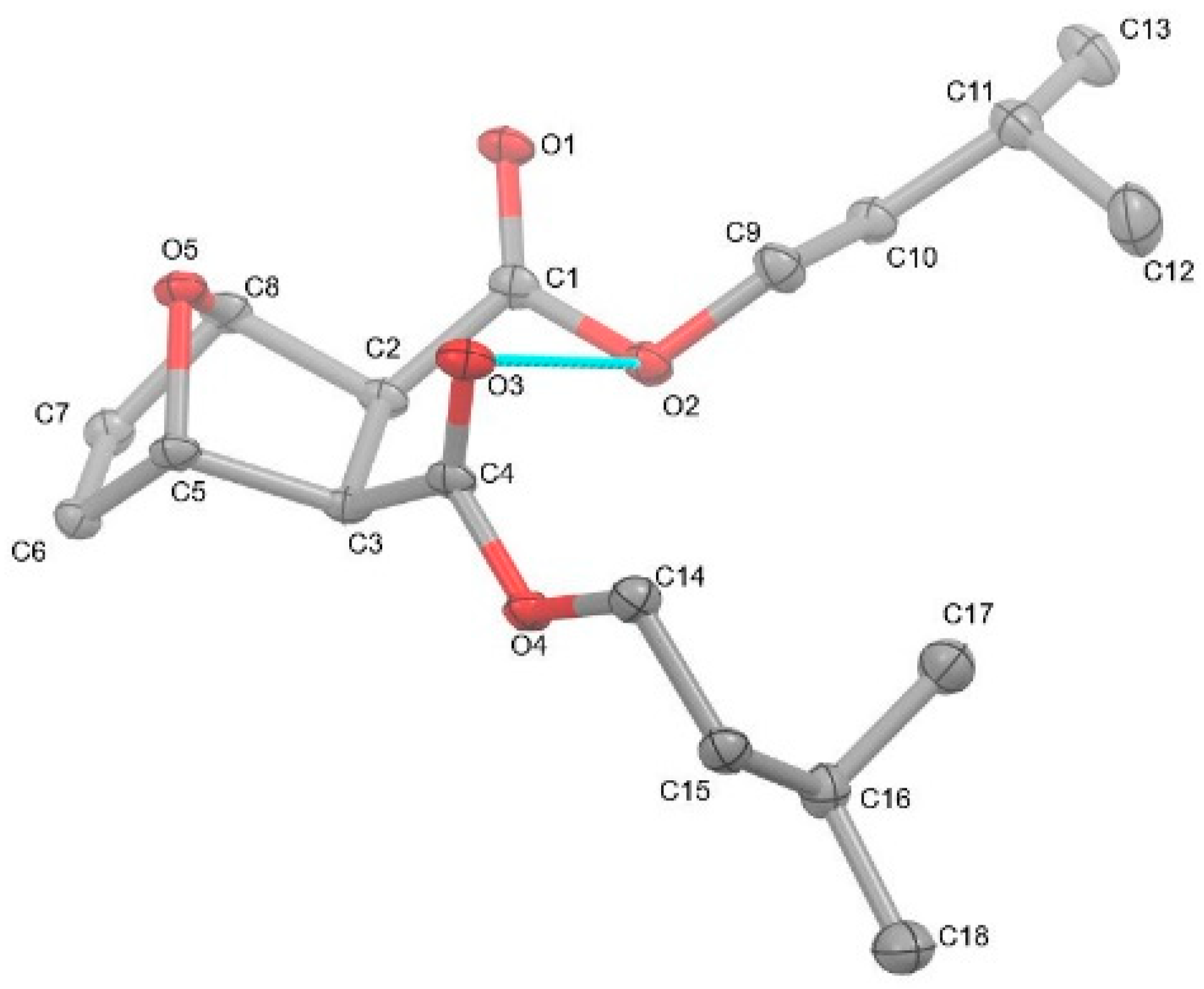
Compound 2 was examined by single-crystal X-ray diffraction (Figure 3). Crystal data for C18H28O5 (MW: 324.19 g/mol): monoclinic, space group P21/c (14) with cell values of a = 15.5647(3) Å, b = 12.8969(2), c = 9.0873(2) Å; β= 99.632(2)° V = 1798.44(6) Å3, Z = 4, T = 104.5 K, 0.521 × 0.275 × 0.08 (thickness, length, width reporting), μ(CuKα) = (λ = 1.54184 Å), Dcalc = 1.198 g/cm3, 18,491 reflections measured (5.76 ≤ 2Θ ≤ 139.618°), to 3363 unique (Rint = 0.0487, Rsigma = 0.0262), which were used in all calculations. The final R1 was 0.0494 (I > 2σ(I)) and wR2 was 0.1441 (all data). Despite the numerous reports of 7-oxanorbornene diesters, a perusal of the literature reveals a paucity of their structural reports [8,9,10,11,12,13,14,15,16,17,18,19,20,21]. More surprisingly, only the methyl version has been reported [18,19,20,21]. Thus, this structural report of compound 2 joins the somewhat rare cohort of structurally characterized compounds. The structure of compound 2 displays a bicyclic 7-oxanorbornene unit with protruding isoamyl groups that dominate the structure. The carbonyl groups are tilted in the same direction, causing the carbonyl oxygen atom (O3) to have close intramolecular contact (2.986 Å) with the O2 atom of the adjacent ester moiety. The tilting of the carbonyls is likely an artifact to lessen the intramolecular electronic interactions between the adjacent diesters’ oxygen atoms (O1···O3 = 3.416 Å and O2···O4 = 3.257 Å). This was also observed with the previously reported dimethyl 7-oxanorbornene diester [18,19,20,21]; however, the carbonyl oxygen atoms were a bit closer (O···O = 3.345 Å) as compared to compound 2. Moreover, the intramolecular contact between the carbonyl oxygen atom of one ester is farther away from the ester oxygen atom of the adjacent ester (3.133 Å) in the dimethyl version as compared to compound 2. Canonically, compound 2 would be expected to be a meso compound with Cs symmetry; however, the titling carbonyl groups and the different rotational conformations of the isoamyl groups in the single-crystal X-ray structure of compound 2 cause the molecule to be asymmetric. The bond distances and angles of compound 2 are typical and similar to previously known 7-oxanorbornene diesters and diacids [18,19,20,21]. A list of structural data is shown in Table 1.
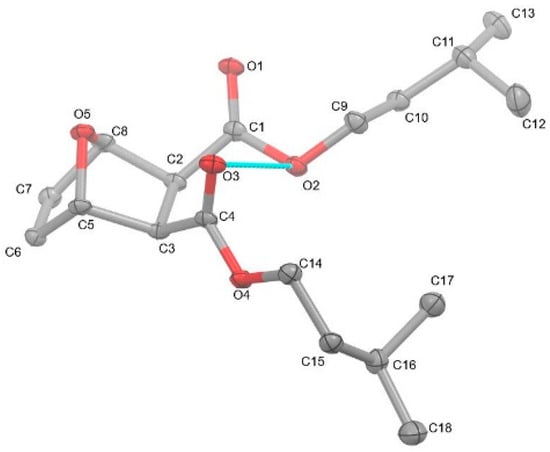
Figure 3.
Single-crystal X-ray structure of diisoamyl (1R, 4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (2). Thermal ellipsoids are shown at 30%. Hydrogen atoms on the carbon atoms have been hidden for clarity.

Table 1.
Geometric parameters (bonds, angles (Å,°)) for compound 2.
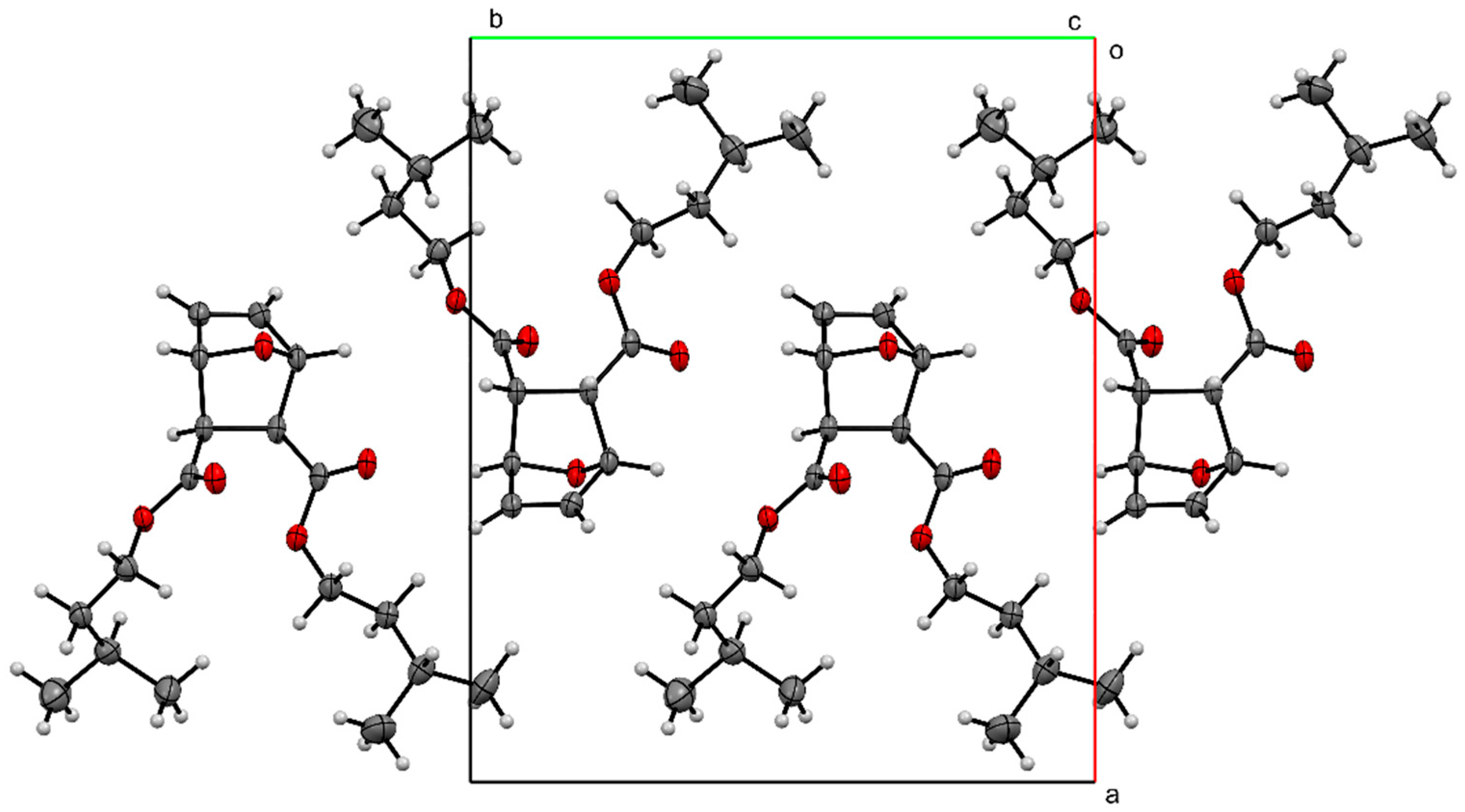
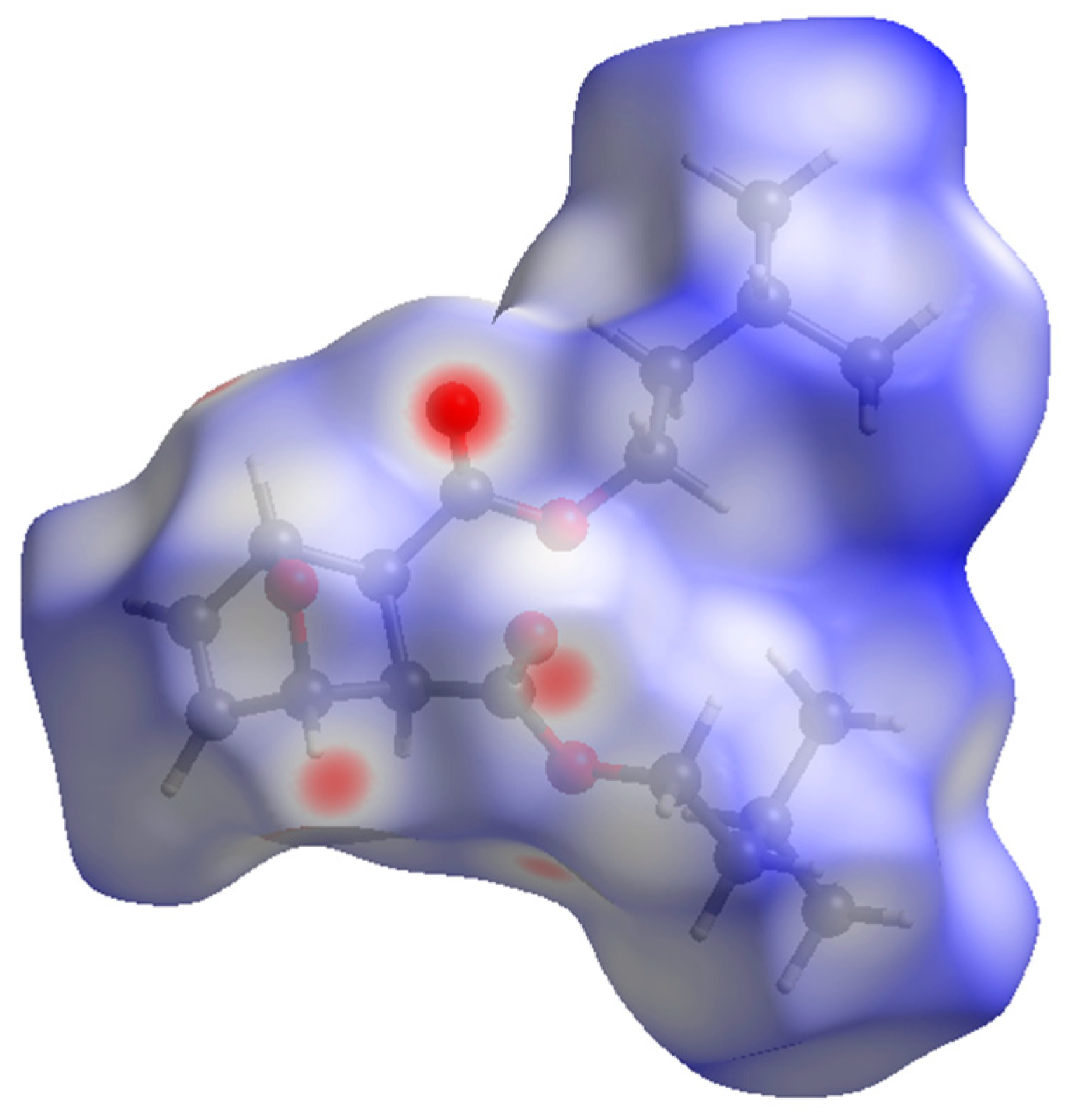
The packing of compound 2 (Figure 4) shows the molecules forming head-to-tail sheets in the a-b plane, which in turn form layers in the c-direction that are stitched together with short contacts between O3…H5–C5 and O1…H2–C2 of neighboring molecules. The intermolecular interactions of compound 2 were further investigated through a quantitative analysis of the Hirshfeld surface, visualized using the Crystal Explorer 21 software package [22]. The Hirshfeld surfaces of the molecules were mapped with the function dnorm (Figure 5). This figure color codes the contacts, with red being shorter than the sums of vdW radii, blue longer than the sums of vdW radii, and white areas approximately equal to vdW. For compound 2, the most intense red spots correspond to the intermolecular contacts between O1 and H2 [2.3029 (11) Å], and between O3 and H5 [2.4384 (11) Å]. Analysis of the two-dimensional fingerprint plot [23] indicates that H…H contacts are the most common in this structure, representing 75.2% of the intermolecular contacts. O…H contacts make the second-highest contribution, representing 21.7% of the contacts. The other types of contacts combined represent less than 10% of the contacts.
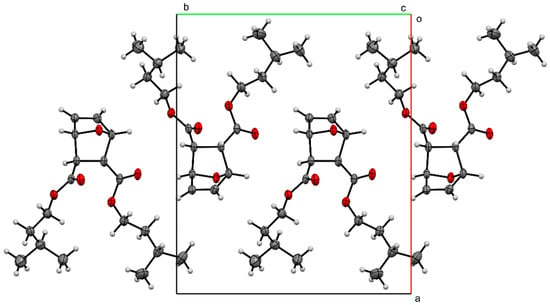
Figure 4.
Packing of compound 2, viewed down the c-axis.
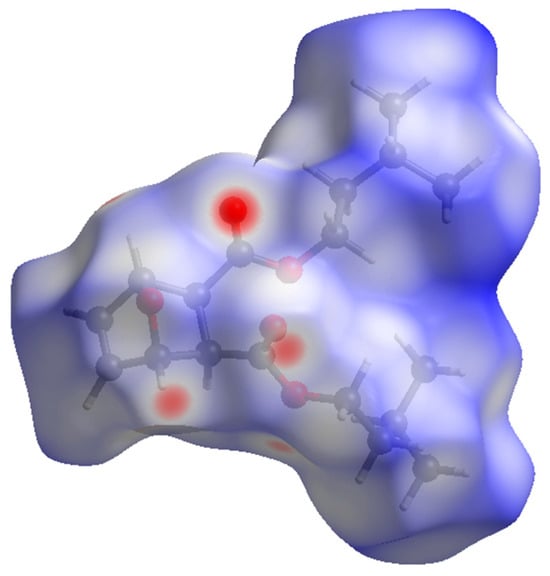
Figure 5.
Hirshfeld surface for compound 2 mapped over dnorm.
2.2. Spectroscopy and Characterization
The 1H and 13C nuclear magnetic resonance spectra of compound 2 correlate well with its structure. The protons on the bicyclic, 7-oxanorbornene scaffold all appear as singlets at 6.49, 5.24, and 2.78 ppm owing to a lack of magnetic spin information exchange from neighboring protons. This is due to improper 3J(H–C–C–H) dihedral angles (ϕ) required for coupling. The Karplus equation states that strong 3J coupling is observed when the ϕ of the H–C–C–H orbitals are parallel (synperiplanar (0-30°)or antiperiplanar (150–180°)). Weak coupling occurs when the ϕ of the H–C–C–H bonds is between 60 and 120° [24]. The protons for the methylene groups (OCH2) attached to the ester oxygen atom are diastereotopic and appear as a complicated multiplet with second-order spin couplings representing the behavior of an ABXY system (4.10 ppm). Though diastereotopic, the other methylene group appears as a simple quartet (1.51 ppm), despite the differing chemical environments of its neighboring protons. The resonances for the remaining protons of the isopropyl fragment are as expected (nonet and doublet, CH(CH3)2). The 13C NMR of compound 2 (see Supplementary Materials) shows the expected eight resonance signals with the carbonyl and alkene carbons appearing far downfield (171.62 and 136.72 ppm, respectively). It would be expected that the tertiary methine carbon would be more downfield that of a methylene group; however, a DEPT-135 experiment revealed the methylene group of the CH2CH(CH3)2 fragment is more downfield (37.33 ppm) than the methine carbon (25.18 ppm). The infrared spectrum of compound 2 shows strong stretches at 2958 (sp2) and 2932 (sp3) cm-1 indicative of alkyl groups. The diagnostic carbonyl stretch occurs at 1737 cm−1. The alkene displays a strong stretch at 1463 cm−1. The stretch at 1189 cm−1 correlates to the C-O stretch of the ester, while the stretch at 1149 cm−1 represents the C-O stretch of the bridging ether unit of the 7-oxanorbornene. The UV-vis spectrum of compound 2 shows a λmax of 240 nm (log ε = 1.84) due to the π–π* transition of the carbonyl and another absorption at 278 nm (log ε = 1.43) due to its n–π* transition. Analysis of compound 2 using GC-MS with electron impact ionization does not show the molecular ion, which is common for long-chain esters due to McLafferty rearrangements and other cleavages [25]. The base peak at m/z = 71 is the isoamyl group fragment. Another prominent fragment at m/z = 117 matches that of a maleic acid unit as a result of a retro-Diels–Alder rearrangement and cleavage of the isoamyl groups. A less prominent fragment with m/z = 187 is likely the intact doubly protonated 7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid. Compound 2 has a melting point of 76–78.2 °C, which is lower than that reported for its dimethyl congener (dimethyl (1R,4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate) (120 °C) [14]. The comparatively lower melting point of compound 2 results from the branched isoamyl group, which lessens intermolecular interactions.
3. Materials and Methods
3.1. General
All chemicals were purchased from commercial sources and used as received unless otherwise noted. Exo-7-oxabicyclo[2.2.1] hept-5-ene-2,3-dicarboxylic anhydride (1) was prepared by literature methods [11]. Ultrasonic reactions were conducted in a Fisher Scientific Fisherbrand® sonicator (Model FUB20LD) ( Thermo Fisher Scientific Inc., Waltham, MA, USA) at a frequency of 40 KHz with a power rating of 400 W. All NMR spectra were recorded on a JEOL ECS300 FT 300 MHz spectrometer(JEOL Ltd., Tokyo, Japan). All 1H and 13C {1H} NMR spectra were referenced against residual signals of the deuterated solvent and chemical shifts were recorded in ppm (δ). Coupling constants (JHH values) were reported in Hz. All UV-vis spectra were recorded on a Cary 100 UV-vis double-beam instrument (Agilent Technologies, Inc. Santa Clara, CA, USA) with a multicellchanger. Stock solutions of the complexes were diluted (10 mg of compound 2 in 3 mL solvent) in chloroform and scanned from 200–350 nm at room temperature. Gas chromatography–mass spectrometry (GC–MS) samples were recorded on a Shimadzu GC-2010 gas chromometer with a quadrupole mass spectrometer (QP2010SE) (Shimadzu Corporation, Kyoto, Japan) using an Agilent 191015–44 column (Agilent Inc., Santa Clara, CA, USA). All GC–MS samples were introduced via split injection with a two-min hold time at 50 °C and a temperature ramp of 15 °C/min over a range of 50–250 °C. Single-crystal X-ray diffraction measurements were performed on a Rigaku XtaLab Synergy-i diffractometer (Rigaku Corporation, Tokyo, Japan) equipped with a Cu micro-focused source at 1.5418 Å and a HyPix Bantam detector (Rigaku Corporation, Tokyo, Japan). Infrared spectra were recorded on a PerkinElmer Spectrum-100 FTIR (PerkinElmer, Shelton, MA, USA) using ATR methods. Melting point determinations were recorded on a Reach Device RD-MP digital melting point apparatus (Reach Devices, LLC., Boulder, CO, USA). Elemental analyses were performed at Atlantic Microlab, Inc., Norcross, GA, USA.
3.2. Synthesis
Compound 1 (250 mg, 1.52 mmol) was placed into a conical vial with isoamyl alcohol (3 mL), H2SO4 (2 drops), and a hydrogel bead (Jelly Marbles®). The vial was capped, placed in the sonicator, and heated at 49 °C for 30 min. The reaction was extracted with a 1.0 M solution of K2CO3 (3 mL) and diethyl ether (10 mL) (3 X). The organic extracts were combined and extracted with brine (6 mL). The organic solution was collected and dried with MgSO4. The solution was filtered, and the filtrate was collected. The filtrate solution was allowed to slowly evaporate at room temperature over a week. Long colorless plate-like crystals formed. The crystals were washed with pentane to remove residual isoamyl alcohol. 0.099 g (20.16%). mp 76–78.2 °C. 1H NMR (300 MHz, CDCl3) δ: 0.91 (12H, d, CH(CH3)2, 3JHH = 7 Hz), 1.51 (4H, q, OCH2CH2-, 3JHH = 7 Hz), 1.67 (2H, non., CH(CH3)2, 3JHH = 7 Hz), 2.78 (2H, s, (CO)CH), 4.12 (4H, m, O-CH2), 5.24 (2H, s, HC-O-CH), 6.45 (2H, s, =CH). 13C NMR (75.57 MHz, CDCl3): δ 171.68 (C=O), 136.78 (C=C), 80.65 (HCOCH), 64.01 (O-CH2) 47.06 (CHC=O), 37.33 (CH2CH(CH3)2), 25.18 (CH(CH3)2), 22.62 (CH(CH3)2). Selected IR bands (ATR-IR, cm−1): 1150 (m), 1189 (m), 1253 (s), 1342 (s), 1392 (s), 1463 (s), 1737 (s), 2869 (s), 2932 (s), 2932 (s), 2958 (s). UV-vis (CHCl3), λmax, nm (log ε): 240 (1.84), 278 (1.43). GC-MS (EI), m/z (% relative intensity, ion): 324.80 (0.01, M+), 187.95 (0.81, O/C(OCCC(C)C)=C/C=C(O)\[O]), 117 (48.60, O=C(O[H])/C=C\C(O[H])=[O+]\ [H]), 99.00 (27.19), 71.00 (100.00, CCC(C)C), 55.00 (30.04, CC([CH2+])=C). Anal. Calc’d for 2 (C19H32O5) Theo.(Found): C 66.64(66.52); H 8.70(8.61).
4. Conclusions
We have prepared diisoamyl (1R,4S)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (2) using a modified Fisher Esterification reaction using sonication and hydrogel beads. The isolation of compound 2 adds to the series of alkyl-derived 7-oxanorbornene diesters and joins the select few that have been fully structurally characterized. Quantitative analysis of the Hirshfeld surface of compound 2 suggests the proton adjacent to the bridging oxygen atom of the 7-oxabicyclo[2.2.1]hept-5-ene unit is sufficiently electron-deficient to undergo electrostatic interactions with the oxygen atoms of the ester groups to template the head-to-tail packing.
Supplementary Materials
The following supporting information can be downloaded, Figure S1: 1H NMR of compound 2 recorded in CDCl3 on a 300 MHz spectrometer; Figure S2: 13C NMR spectrum of compound 2 recorded in CDCl3 on a 75.75 MHz spectrometer; Figure S3: A comparison of the 13C NMR spectrum of compound 2 and its DEPT-135 spectrum recorded in CDCl3 on a 75.75 MHz spectrometer; Figure S4: Infrared spectrum of compound 2 (ATR); Figure S5: UV-vis spectrum of compound 2 recorded in CHCl3; Figure S6: GC-MS chromatograph (top) and chromatogram (bottom) of compound 2; Table S1: Crystal data and structure refinement for compound (2); Table S2: Fractional atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103) for compound (2); Table S3: Anisotropic displacement parameters (Å2 × 103) for compound (2). The anisotropic displacement factor exponent takes the form −2π2[h2a*2U11+2hka*b*U12+…]; Table S4: Bond lengths for compound (2); Table S5: Bond angles for compound (2); Table S6: Torsion angles for compound (2); Table S7: Hydrogen atom coordinates (Å × 104) and isotropic displacement parameters (Å2 × 103) for compound (2).
Author Contributions
Conceptualization, B.Q.; methodology, B.Q.; formal analysis, B.Q., K.M., E.M.V., J.G.B., A.R.M. and C.W.P.; investigation, K.M., E.M.V., J.G.B., A.R.M. and C.W.P.; data curation, B.Q.; writing—original draft preparation, K.M. and B.Q.; writing—review and editing, B.Q., K.M., E.M.V., J.G.B., A.R.M. and C.W.P.; visualization, B.Q. and C.W.P.; supervision, B.Q.; project administration, B.Q.; funding acquisition, B.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Foundation Major Research Instrumentation fund, NSF-MRI 2215812, and the National Science Foundation Improving Undergraduate STEM Education: Directorate for STEM Education NSF-IUSE DUE 1611988.
Data Availability Statement
The original contributions presented in this study are included in this article/Supplementary Materials; further inquiries can be directed to the corresponding author/s. Final atomic coordinates, geometrical parameters, and crystallographic data have been deposited to the Cambridge Crystallographic Data Centre, 11 Union Road, Cambridge, CB2 1EZ, UK (https://www.ccdc.cam.ac.uk/structures/) and are available on request quoting the deposition number CCDC 2363397.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of this study; in the collection, analyses, or interpretation of data in the writing of this manuscript; or the decision to publish the results.
References
- McMurry, J. Fundamentals of Organic Chemistry; Brooks/Cole: Monterey, CA, USA, 1986. [Google Scholar]
- Khan, Z.; Javed, F.; Shamair, Z.; Hafeez, A.; Fazal, T.; Aslam, A.; Zimmerman, W.B.; Rehman, F. Current developments in esterification reaction: A review on process and parameters. J. Ind. Eng. Chem. 2021, 103, 80–101. [Google Scholar] [CrossRef]
- Trnka, T.M.; Grubbs, R.H. The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Ulman, M.; Grubbs, R.H. Relative Reaction Rates of Olefin Substrates with Ruthenium(II) Carbene Metathesis Initiators. Organometallics 1998, 17, 2484–2489. [Google Scholar] [CrossRef]
- Grubbs, R.B.; Grubbs, R.H. 50th Anniversary Perspective: Living Polymerization—Emphasizing the Molecule in Macromolecules. Macromolecules 2017, 50, 6979–6997. [Google Scholar] [CrossRef]
- Novak, B.M.; Grubbs, R.H. The Ring-Opening Metathesis Polymerization of 7-Oxabicyclo[2.2.1]Hept-5-Ene Derivatives—A New Acyclic Polymeric Ionophore. J. Am. Chem. Soc. 1988, 110, 960–961. [Google Scholar] [CrossRef]
- Novak, B.M.; Grubbs, R.H. Catalytic Organometallic Chemistry in Water: The Aqueous Ring-Opening Metathesis Polymerization of 7-Oxanorbornene Derivatives. J. Am. Chem. Soc. 1988, 110, 7542–7543. [Google Scholar] [CrossRef]
- Holerca, M.N.; Peterca, M.; Partridge, B.E.; Xiao, Q.; Lligadas, G.; Monteiro, M.J.; Percec, V. Monodisperse Macromolecules by Self-Interrupted Living Polymerization. J. Am. Chem. Soc. 2020, 142, 15265–15270. [Google Scholar] [CrossRef]
- deRonde, B.M.; Posey, N.D.; Otter, R.; Caffrey, L.M.; Minter, L.M.; Tew, G.N. Optimal Hydrophobicity in Ring-Opening Metathesis Polymerization-Based Protein Mimics Required for siRNA Internalization. Biomacromolecules 2016, 17, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Kyu, A.J.; Lim, K.H.; Chul, S.K.; Hoon, L.S. Catalysts for Polymerization or Copolymerization of Olefins, Its preparing Method and Olefins Polymerization or Copolymerization Method. KR20120077690A, 10 July 2012. [Google Scholar]
- Karlen, T.; Ludi, A.; Mühlebach, A.; Bernhard, P.; Pharisa, C. Photoinduced ring opening metathesis polymerization (PROMP) of strained bicyclic olefins with ruthenium complexes of the type [(η6-arene1)Ru(η6-arene2)]2+ and [Ru(Nc-R)6]2+. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 1665–1674. [Google Scholar] [CrossRef]
- Kotsuki, H.; Nishizawa, H.; Ochi, M.; Matsuoka, K. High Pressure Organic Chemistry. V. Diels-Alder Reactions of Furan with Acrylic and Maleic Esters. Bull. Chem. Soc. Jpn. 2006, 55, 496–499. [Google Scholar] [CrossRef]
- Baggio, S.; Barriola, A.; de Perazzo, P.K. Crystal and molecular structure of 7-oxabicyclo[2,2,1]hept-5-ene-2,3-exo-dicarboxylic anhydride. J. Chem.Soc. Perkin Trans. 2 1972, 934–937. [Google Scholar] [CrossRef]
- France, M.B.; Alty, L.T.; Earl, T.M. Synthesis of a 7-oxanorbornene derivative: A two-step sequence preparation for the organic laboratory. J. Chem. Educ. 1999, 76, 659–660. [Google Scholar] [CrossRef]
- Li, J.J. (Ed.) Fischer–Speier esterification. In Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications Fifth Edition; Springer International Publishing: Cham, Switzerland, 2014; p. 252. [Google Scholar]
- Martínez, R.F.; Cravotto, G.; Cintas, P. Organic Sonochemistry: A Chemist’s Timely Perspective on Mechanisms and Reactivity. J. Org. Chem. 2021, 86, 13833–13856. [Google Scholar] [CrossRef]
- Flannigan, D.J.; Suslick, K.S. Inertially confined plasma in an imploding bubble. Nat. Phys. 2010, 6, 598–601. [Google Scholar] [CrossRef]
- Miró Vera, A.; Velásquez, W.; Briceño, A.; Bahsas Bahsas, A.; Ramírez Valero, B.; Diaz de Delgado, G. Synthesis and Crystal Structure of Dimethyl-7-oxabicyclo[2.2.1]hept-5-ene exo,exo-2,3-dicarboxylate. J. Chem. Crystallogr. 2007, 37, 543–548. [Google Scholar] [CrossRef]
- Li, J. Benzothiazol-2-amine-3-methoxycarbonyl-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylic acid (1/1). Acta Crystallogr. Sect. E 2011, 67, o199. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Goh, R.Y.W. CCDC 1027454. CSD Communication 2014. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/cc13h4pf&sid=DataCite (accessed on 11 July 2024). [CrossRef]
- Sadeghi-Khomami, A.; Blake, A.J.; Wilson, C.; Thomas, N.R. Synthesis of a Carbasugar Analogue of a Putative Intermediate in the UDP-Galp-Mutase Catalyzed Isomerization. Org. Lett. 2005, 7, 4891–4894. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Minch, M.J. Orientational dependence of vicinal proton-proton NMR coupling constants: The Karplus relationship. Concepts Magn. Reson. 1994, 6, 41–56. [Google Scholar] [CrossRef]
- McLafferty, F.W. Mass Spectrometric Analysis. Molecular Rearrangements. Anal. Chem. 1959, 31, 82–87. [Google Scholar] [CrossRef]
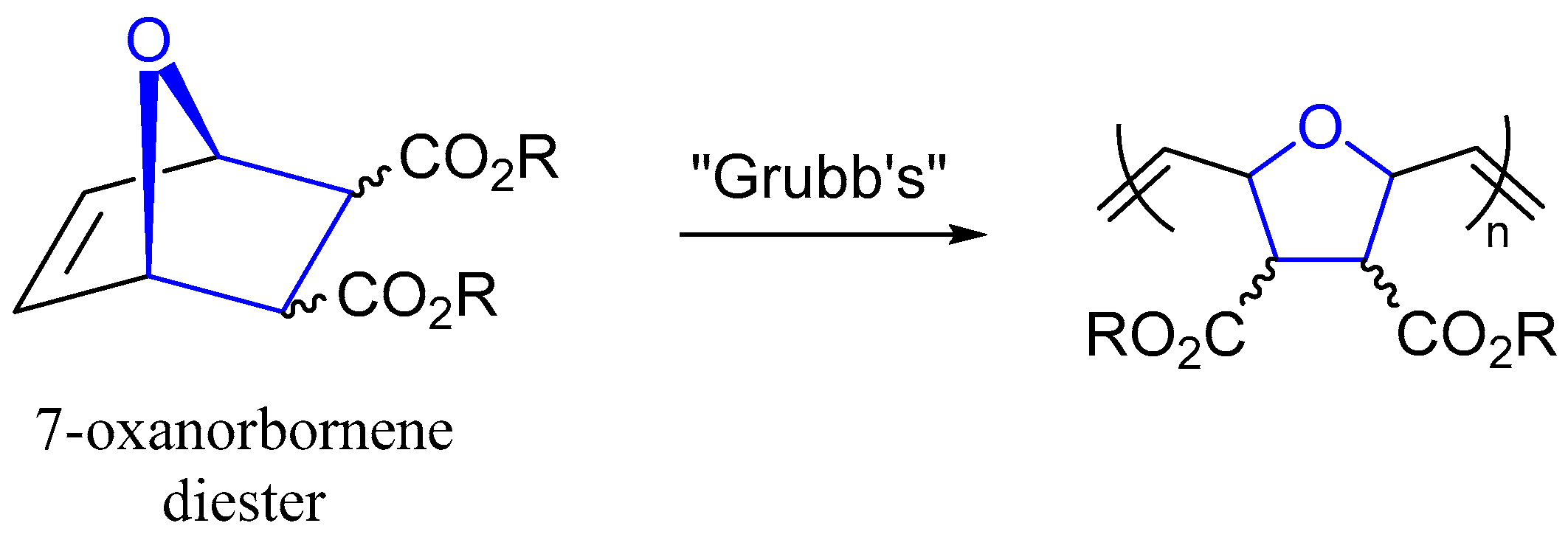
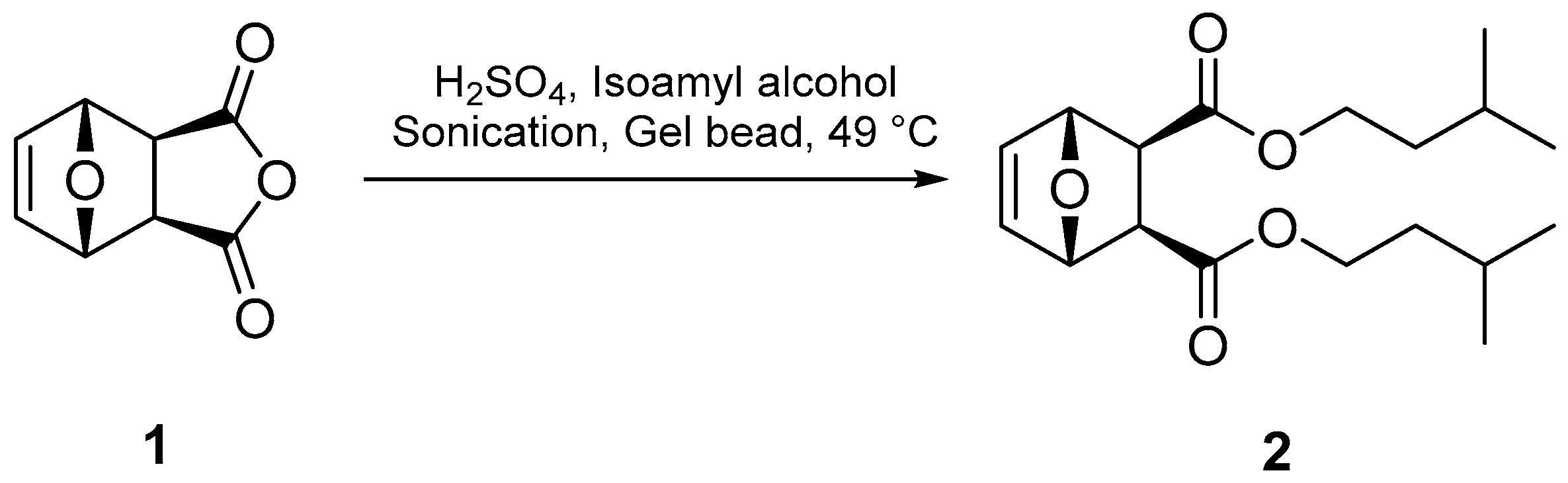
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).