
(E)-1-(2,5-Dimethylphenyl)-3-phenylprop-2-en-1-one



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
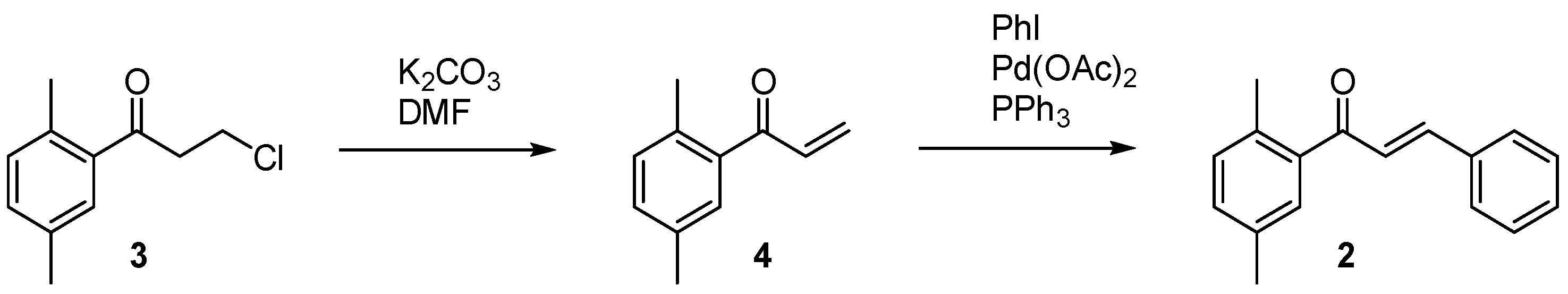
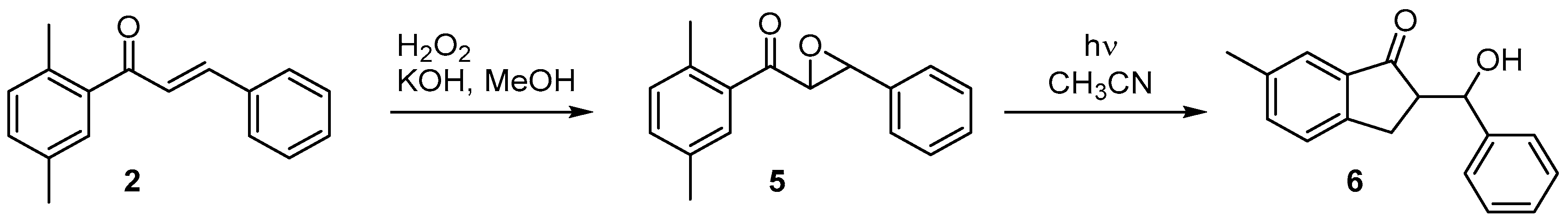
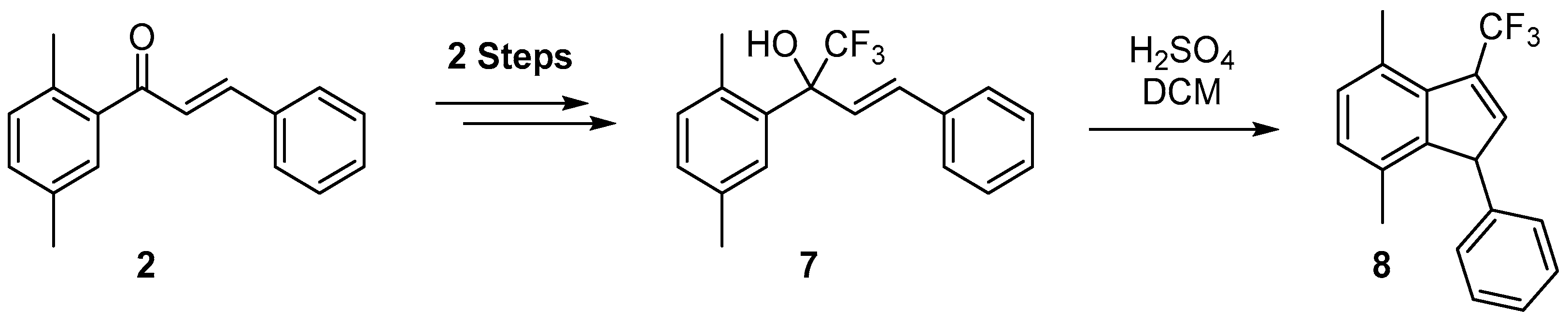
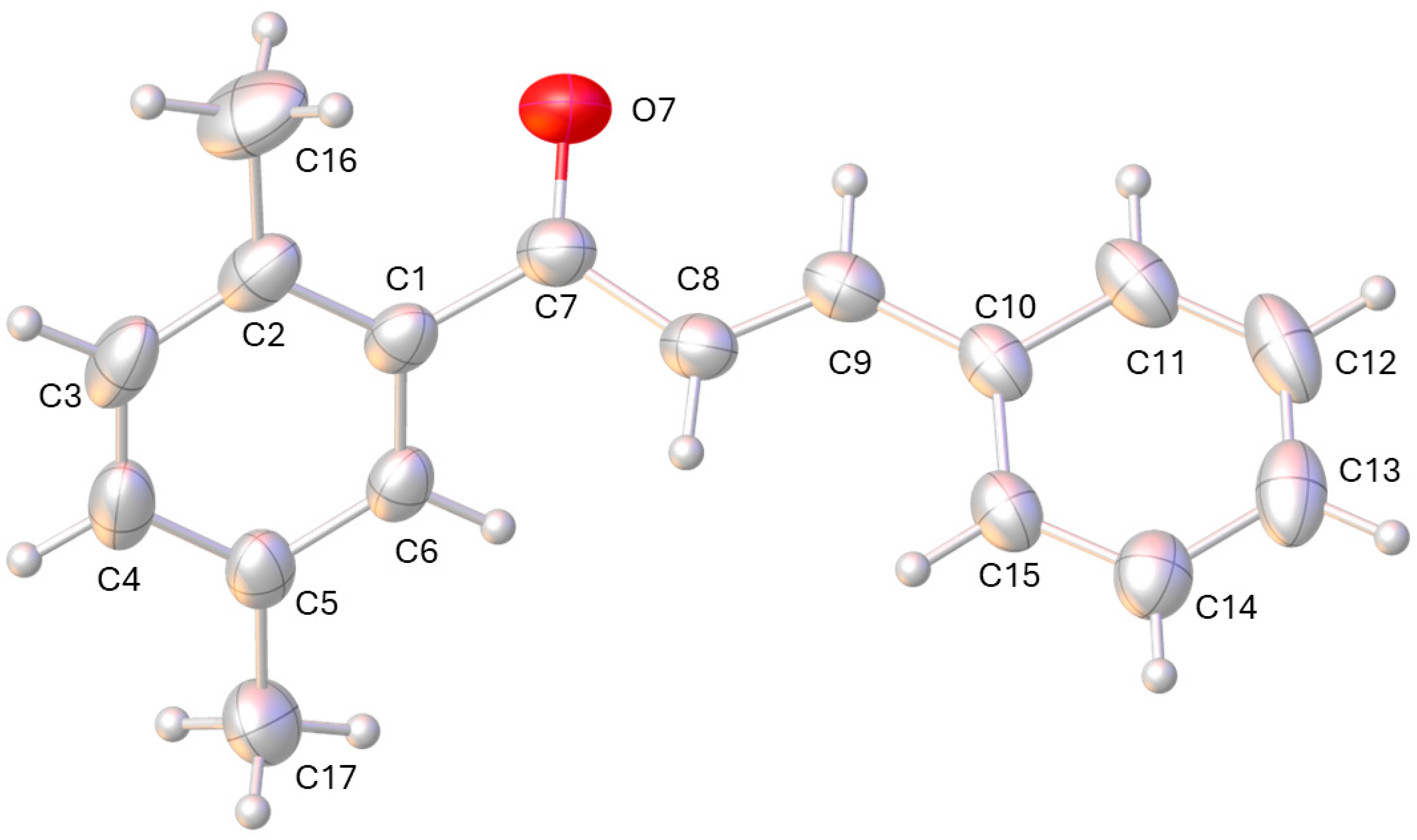
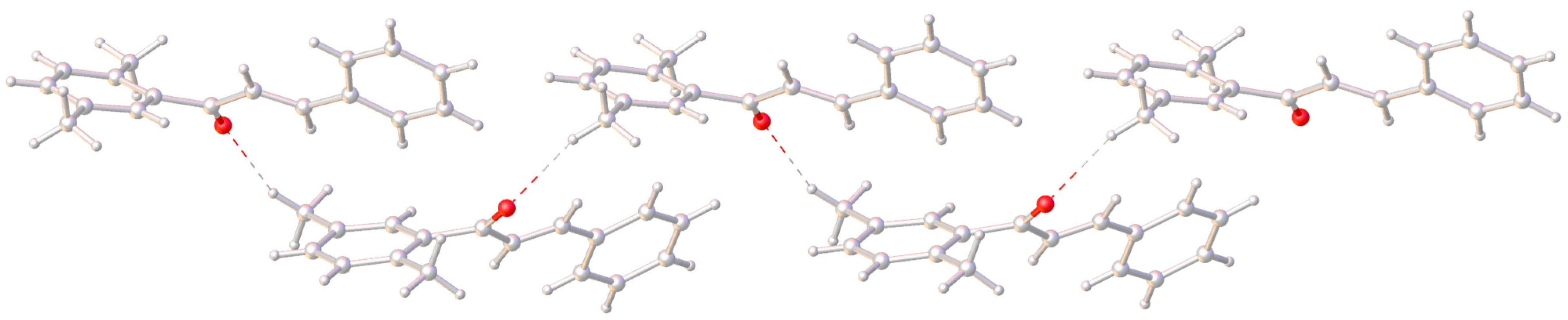
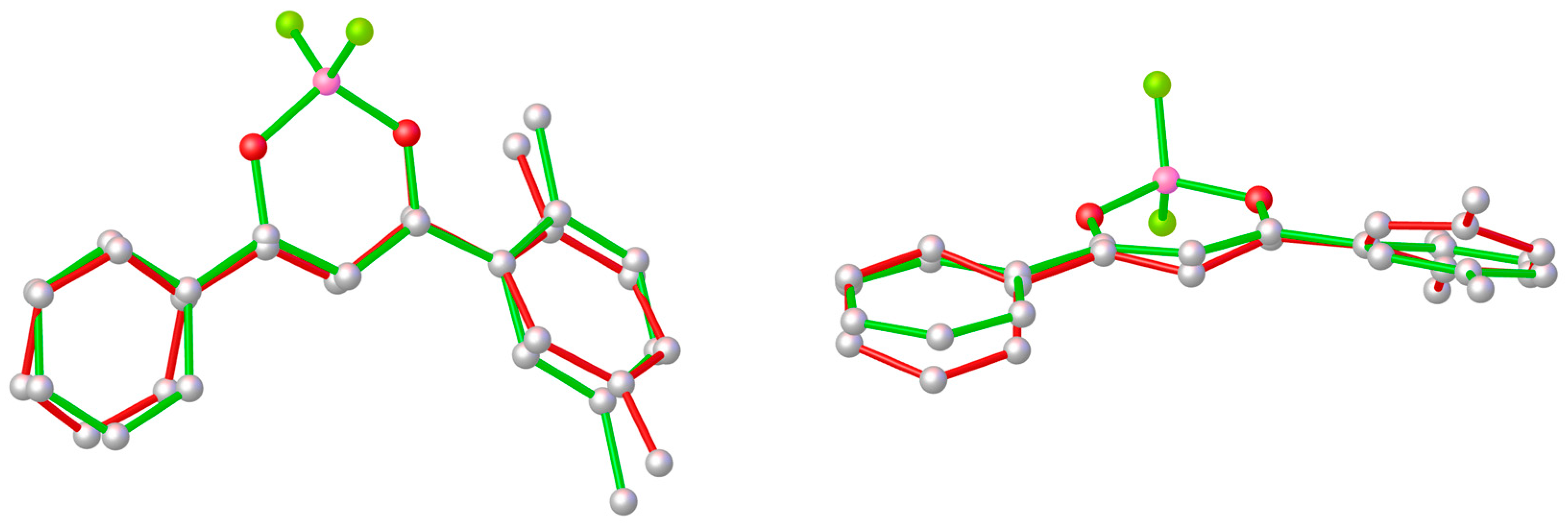
2. Results
3. Materials and Methods
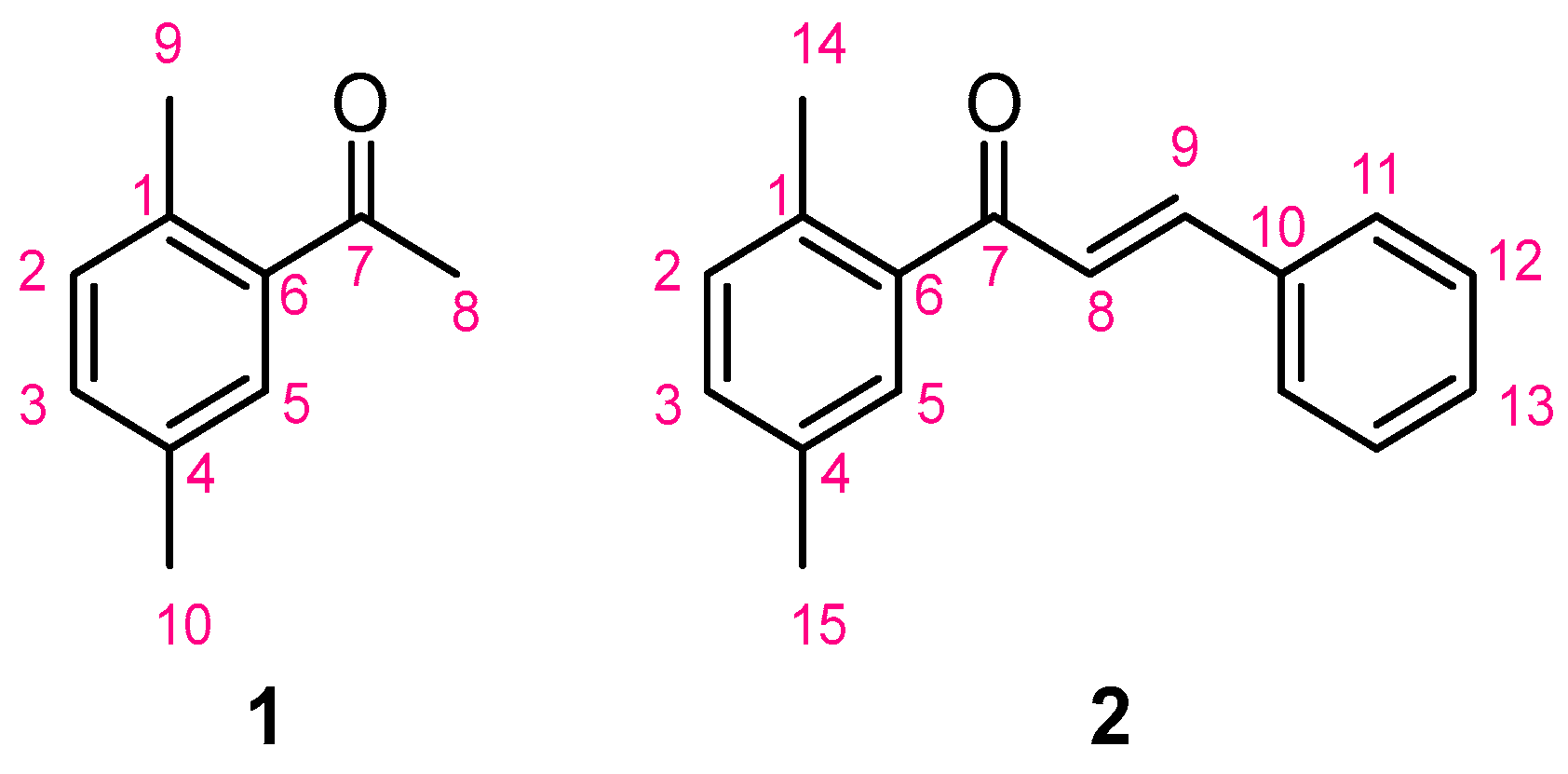
3.1. Synthesis of 1-(2,5-Dimethylphenyl)ethan-1-one (1)
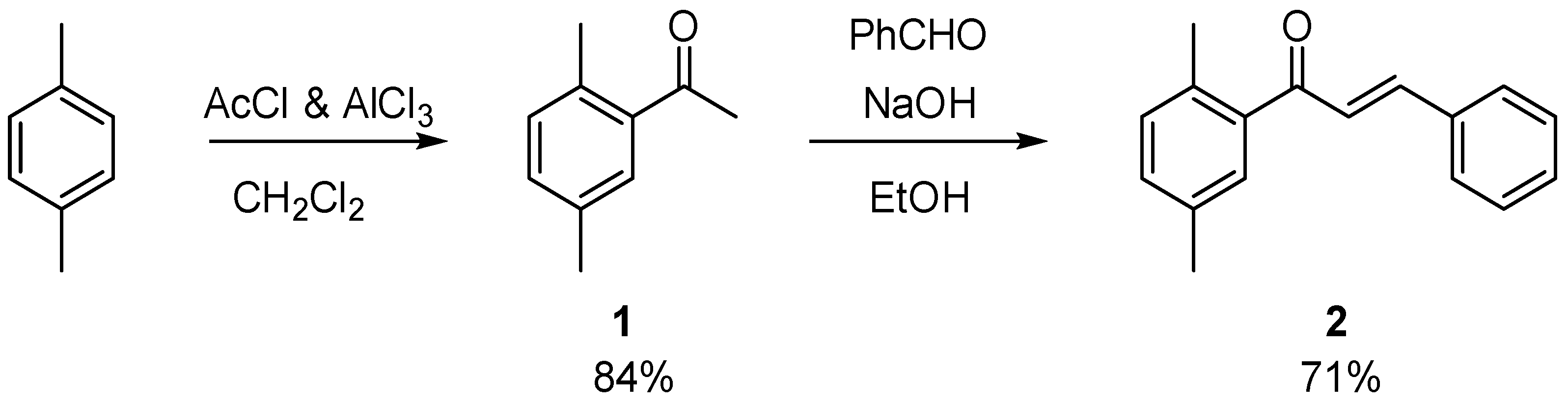
3.2. Synthesis of 1-(2,5-Dimethylphenyl)-3-phenylprop-2-en-1-one (2)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Solomek, T.; Stacko, P.; Veetil, A.T.; Pospíšil, T.; Klán, P. Photoenolization-Indued Oxirane Ring Opening in 2,5-Diemethylbenzoyl Oxiranes to form Pharmaceutically Promising Indanone Derivatives. J. Org. Chem. 2010, 75, 7300–7309. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Han, C.; Xu, J. A green synthesis approach toward large-scale production of benzalacetone via Claisen-Schmidt condensation. RSC Adv. 2022, 12, 29240–29245. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Jiang, Q.; Yu, L.; Yu, Z. Synthesis of chalcones via domino dehydrochlorination/Pd(OAc)2-catalyzed Heck reaction. Chin. J. Catal. 2015, 36, 78–85. [Google Scholar] [CrossRef]
- Martynov, M.Y.; Iakovenko, R.O.; Kazakova, A.N.; Boyarskaya, I.A.; Vasilyev, A.V. Acid-promoted cyclisation of 2,3-diaryl-1,1,1-trifluorobut-3-en-2-oles and their TMS-ethers into CF3-indenes. Org. Biomol. Chem. 2017, 15, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, T.; Veetil, A.T.; Antony, L.A.P.; Klán, P. Photochemical synthesis of substituted indan-1-ones related to donepezil. Photochem. Photobiol. Sci. 2008, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Rabinovich, D. Topochemistry. Part XXX. Crystal and molecular structures of chalcone. J. Chem. Soc. B 1970, 11–16. [Google Scholar] [CrossRef]
- Tanaka, M.; Muroka, S.; Matsui, Y.; Ohta, E.; Ogaki, T.; Mizuno, K.; Ikeda, H. Cooperative effects of o- and m-methyl groups on the intramolecular charge−transfer emission properties of dibenzoylmethanato boron difluorides. Photochem. Photobiol. Sci. 2017, 16, 845–853. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro, v1.171.43.109a; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2024.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordes, D.B.; Smellie, I.A.; Chalmers, B.A. (E)-1-(2,5-Dimethylphenyl)-3-phenylprop-2-en-1-one. Molbank 2024, 2024, M1862. https://doi.org/10.3390/M1862
Cordes DB, Smellie IA, Chalmers BA. (E)-1-(2,5-Dimethylphenyl)-3-phenylprop-2-en-1-one. Molbank. 2024; 2024(3):M1862. https://doi.org/10.3390/M1862
Chicago/Turabian StyleCordes, David B., Iain A. Smellie, and Brian A. Chalmers. 2024. "(E)-1-(2,5-Dimethylphenyl)-3-phenylprop-2-en-1-one" Molbank 2024, no. 3: M1862. https://doi.org/10.3390/M1862
APA StyleCordes, D. B., Smellie, I. A., & Chalmers, B. A. (2024). (E)-1-(2,5-Dimethylphenyl)-3-phenylprop-2-en-1-one. Molbank, 2024(3), M1862. https://doi.org/10.3390/M1862