Abstract
A general protocol for the synthesis of 3,4-dihalogen substituted 1,8-naphthalimides is proposed, starting from available and cheap 1,8-naphthalic anhydride. The reported new compounds have only bromine or chlorine atoms as substituents, in contrast to the known iodo-containing analogues. This is an advantage in possible aryl nucleophilic substitution or cross-coupling modifications, making them interesting and important building-block molecules in naphthalimide chemistry. Although the procedure includes five synthetic steps, they are quick and straightforward. The overall yields are relatively high (48–62%), and only one column of chromatographic purification is needed. All the reactions were carried out on a multigram scale to allow the target building-block compounds to be obtained in sufficient amounts for further derivatizations.
1. Introduction
Naphthalimides (NIs) are heterocyclic compounds with a planar, π-deficient aromatic core structure. Their derivatives, especially having electron-donating substituents, are characterized by valuable spectral properties, as well as promising biological activities [1,2]. Numerous applications of 1,8-naphthalimide derivatives in various research fields have been reported in the literature in the last decades. These versatile molecules were successfully exploited as fluorescent chemosensors for monitoring biologically important cations, anions, and enzymes [3,4], probes for the detection of small molecular substances in the environment and in vivo [5], sensitizers [6], anticancer agents, DNA intercalators [7,8], emissive layers in OLEDs [9], organelle imaging and tracking agents [10], etc. Despite the constant search for new NI-based compounds with tailored properties, the synthetic methods for derivatization of the core structure, either in the imides or in their anhydride precursors, remain limited. In most cases, halogen-substituted starting compounds are used since they can undergo aromatic nucleophilic substitution and/or metal-catalyzed cross-coupling reactions to afford the target NIs of interest. Unfortunately, aside from 4-halogeno substituted ones, they are not commercially available and must be prepared in advance, which might become challenging, especially for poly-halogen-containing representatives. From that point of view, the development of easily accessible synthetic protocols for preparation on the gram scale of polyhalogenated naphthalic anhydrides and their respective imides is greatly beneficial for rylene chemistry.
Recently, we reported a method for excessive halogenation of naphthalic anhydride to prepare 3,4,5,6-tetrabromo and tetrachloro derivatives, which were proven as versatile building blocks for further derivatization [11,12]. A more selective method for the synthesis of 3,4,6-tribromo naphthalic anhydride was later developed to allow new substitution patterns to be exploited [13]. As part of our efforts to expand the toolbox of synthetic chemists in the field, we focused on improving strategies for preparing 3,4-dihalogeno NIs.
The first example of a synthesis of such a compound is the work of Brana et al. [14], where the authors obtained 4-chloro-3-iodo NI, which was then used in Sonogashira cross-coupling. More than a decade later, in the group of Professor Tian, 4-bromo-3-iodo NI was synthesized [15], and both halogens were substituted with thiophene units in Suzuki cross-coupling. The same approach was used later again by Tian et al. [16] and by the group of Liu and Yin [17,18]. Most recently, Kulik et al. synthesized 3-chloro-4-iodo NI from 3-amino naphthalic anhydride, which was then used for the preparation of aryl-bridged thienonaphthalimides [19]. In all cases, the second halogen is introduced under Sandmeyer reaction conditions. In all examples, one of the halogens is iodine, which leads to two significant disadvantages: (i) the total yields are not very high, and (ii) the iodo-NIs are not the ideal precursors neither for aromatic nucleophilic substitution nor for metal-catalyzed cross-coupling reactions, due to significant amounts of deiodination. To tackle these problems, we have developed and report herein an improved strategy for gram-scale preparation of 3,4-dichloro or 3,4-dibromo-NI, as well as mixed 3,4-dihalogenated bromo-chloro naphthalimides, starting from cheap and commercially available 1,8-naphthalic anhydride.
2. Results and Discussion
The lack of commercially available and inexpensive polysubstituted 1,8-naphthalic anhydrides or easily accessible synthetic protocols for their preparation greatly limits the possibility of synthesizing diverse polyfunctional NI derivatives. As our literature survey showed (vide supra), there are only a few examples of preparing 3,4-dihalogen substituted 1,8-naphthalimides, and all reported methods use 3-amino-1,8-naphthalene anhydride. The use of substituted 1,8-naphthalic anhydrides in the conditions of the Sandmeyer reaction has several disadvantages: (i) low solubility of the anhydrides in aqueous acidic media or organic solvents; (ii) inability to purify the product by column chromatography or recrystallization; (iii) difficulties in following the reaction by TLC due to the strong retention on silica gel, as well as the easy opening of the cyclic anhydride to a dicarboxylic acid. To avoid these drawbacks, we decided to use an orthogonal approach for the synthesis of 3,4-dihalogen substituted NIs based on N-alkyl imides instead of anhydrides (Scheme 1).
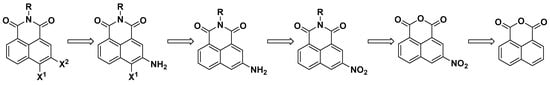
Scheme 1.
Synthetic strategy for preparation of 3,4-dihalogen 1,8-naphthalimides.
Our synthetic strategy involves several standard steps used in naphthalimide chemistry. It starts with nitration of the cheap and commercially available 1,8-naphthalic anhydride, followed by imidization, reduction of 3-nitro-1,8-naphthalimide to an amino derivative, and halogenation under mild conditions. The key step was finding suitable conditions for the Sandmeyer reaction of the significantly hydrophobic N-alkyl-1,8-naphthalimide to proceed.
Based on our previous research, we carried out nitration in concentrated sulfuric acid and solid sodium nitrate at room temperature (Scheme 2). The 3-nitro-1,8-naphthalic anhydride obtained under these conditions contains small amounts of the isomeric 4-nitro-1,8-naphthalic anhydride, but it is easily removed with just one recrystallization from chlorobenzene. The imidization of 3-nitro-1,8-naphthalic anhydride 2 with 2-ethylhexylamine was carried out in a mixture of NMP and acetic acid at 120 °C for 30 min. After workup, the crude product (pure by TLC) was dissolved in a minimal amount of dichloromethane, and methanol was added portion-wise until imide 3 was completely precipitated. For the reduction, various conditions were tested—palladium on carbon and hydrogen or hydrazine, stannous dichloride or iron powder in acetic acid, or a mixture of dioxane and concentrated hydrochloric acid (see the Supplementary Information for details). The best results were obtained using iron powder in acetic acid at 90 °C. Under these conditions, the reduction proceeds relatively quickly and very neatly and is easily followed by TLC. After workup, the crude product was dissolved in dichloromethane, filtered through silica gel, washed with dichloromethane ethyl acetate (20:1), and evaporated to dryness. The target product was isolated in very high yields (99%) on a large scale (100 mmol).
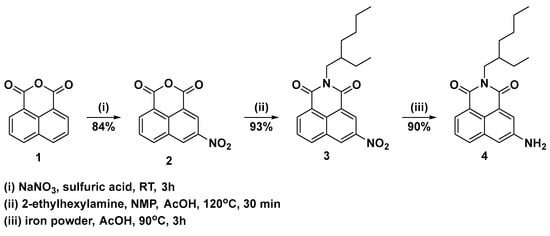
Scheme 2.
Synthesis of N-(2-etylhexyl)-3-amino-1,8-naphthalimide 4.
Since 3-amino-1,8-naphthalimide 4 is a highly activated aromatic substrate for electrophilic aromatic substitution, very mild conditions for the halogenation may be used. The bromination of imide 4 was carried out in acetic acid with 1.2 equivalents of bromine at room temperature (Scheme 3). The reaction is very fast and takes place in only 20 min, isolating the 3-amino-4-bromo NI 5 in quantitative yield and high purity on a gram scale. The chlorination of imide 4 was again carried out by an electrophilic mechanism, using N-chlorosuccinimide in DMF at room temperature. The reaction was completed for five hours, and after workup, the 3-amino-4-chloro imide 6 was isolated in a quantitative yield and pure by TLC.
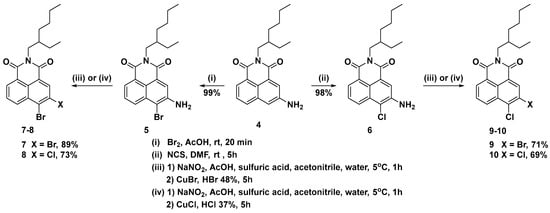
Scheme 3.
Synthesis of 3,4-dihalogen-1,8-naphthalimides.
As mentioned above, the key step in our strategy was to find suitable conditions to carry out the Sandmeyer reaction using the significantly hydrophobic imides 5 and 8. As solvent for the diazotization reaction, we chose acetic acid or a mixture of acetic acid and acetonitrile (2:1), in the presence of 70% aqueous sulfuric acid. In both cases, the diazotization reaction proceeded successfully, with significantly better results observed when the acetonitrile additive was used. After diazotization, the resulting diazonium salt is poured onto a solution of copper bromide or chloride in concentrated hydrobromic or hydrochloric acid, respectively. The yields of the Sandmeyer halogenations carried out in this way were good to excellent (69–89%), and in all cases, the reactions were carried out on a large scale (40 mmol). The use of imides instead of anhydrides in this reaction has the additional advantage of isolating the target 3,4-dihalogenated NIs in high purity by means of column chromatography on silica.
3. Materials and Methods
3.1. Materials
All starting materials and solvents were commercially available and used without additional purification: Fluorochem (Glossop, UK), Across (Antwerpen, Belgium), and Fisher Scientific (Hampton, NH, USA).
NMR spectra were recorded on a Bruker Avance 500 MHz instrument (Bruker, Karlsruhe, Germany) operating at 500 and 126 MHz for 1H and 13C, respectively. CDCl3 was used as a solvent. Chemical shifts are reported in δ units (ppm) and referenced to the residual solvent signals (1H at 7.26 ppm and 13C at 77.160 ppm). HRMS were recorded on a ThermoFisher Scientific—Orbitrap Exploris 120 (Source—HESI APCI, Comby Nozzle, Bremen, Germany). IR spectra were recorded on a Spectrum Two FT-IR spectrometer (PerkinElmer, Waltham, MA, USA) equipped with an ATR accessory. The melting point (uncorrected) was determined on a Kofler hot-stage microscope. Elemental analyses were carried out on a Leco CHNS-932 (Leco Europe, Geleen, The Netherlands). Thin layer chromatographic (TLC) analysis was performed on silica gel plates (Macherey-Nagel F60 254 40 × 80; 0.2 mm, Macherey-Nagel, Duren, Germany).
3.2. Synthesis of 3-Nitro-1,8-naphthalic Anhydride 2
To a solution of 1,8-naphthalic anhydride (200.0 mmol, 39.64 g) in 250 mL conc. sulfuric acid, sodium nitrate (210.0 mmol, 14.49 g) was added in small portions (~500 mg) for two hours at room temperature. The reaction mixture was stirred for an additional one hour at the same temperature. The mixture was then poured onto ice, and the precipitate was filtered, washed with water, and dried. The crude product was purified by recrystallization from chlorobenzene. Yield 40.85 g (84%) as an off-white solid.
3.3. Synthesis of N-(2-Ethylhexyl)-3-nitro-1,8-naphthalimide 3
To a solution of 3-nitro-1,8-naphthalic anhydride 2 (150.0 mmol, 36.48 g) in a 150 mL mixture of NMP and acetic acid (ratio 1:1), 2-ethylhexylamine (1.5 eq, 225.0 mmol, 29.08 g, 37.20 mL) was added. The mixture was stirred for 30 min at 120 °C and then poured into a mixture of ice (200 g) and 50 mL hydrochloric acid. The precipitate was filtered, washed with water, and dried. The crude product was dissolved in dichloromethane, filtered through silica (50 g), washed with dichloromethane, and evaporated to dryness. The solid was dissolved again in a minimum amount of dichloromethane and precipitated with methanol. Yield 49.44 g (93%) as an off-white solid. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 9.31 (1H, d, J = 2.2 Hz); 9.12 (1H, d, J = 2.2 Hz); 8.77 (1H, dd, J = 7.3, 1.1 Hz); 8.42 (1H, m, J = 8.3, 1.0 Hz); 7.94 (1H, dd, J = 8.3, 7.3 Hz,); 4.14 (2H, qd, J = 12.9, 7.3 Hz); 1.93 (1H, hept, J = 6.2 Hz); 1.44–1.26 (8H, m); 0.93 (3H, t, J = 7.4 Hz); 0.88 (3H, t, J = 7.1 Hz).13C NMR (Chloroform-d, 126 MHz) δ ppm: 163.61, 162.99, 146.56, 135.56, 134.59, 131.14, 130.36, 129.24, 128.94, 124.89, 124.43, 123.41, 44.67, 38.05, 30.84, 28.78, 24.16, 23.18, 14.22, 10.73. FT-IR νmax 1704, 1661, 1547, 1347, 1327, 791, 758 cm−1. Anal. calcd. C20H22N2O4: C, 67.78; H, 6.26; N, 7.90; found: C, 67.79; H, 6.57; N, 7.77.
3.4. Synthesis of N-(2-Ethylhexyl)-3-amino-1,8-naphthalimide 4
To a hot solution (90 °C) of N-(2-ethylhexyl)-3-nitro-1,8-naphthalimide 3 (120.0 mmol, 42.53 g) in 400 mL acetic acid, iron powder (360.0 mmol, 20.10 g) was added portion-wise for three hours. The mixture was poured into ice/water (400 g), and the precipitate was filtered, washed with water, and dried. The crude product was dissolved in dichloromethane, filtered through silica (50 g), washed with dichloromethane ethyl acetate (20:1), and evaporated to dryness. The solid was dissolved again in a minimum amount of dichloromethane and precipitated with hexane. Yield 35.04 g (90%) as yellow crystals, mp: 141–143 °C. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 8.31 (1H, dd, J = 7.3, 1.0 Hz); 8.05 (1H, d, J = 2.1 Hz); 7.92 (1H, d, J = 8.1 Hz); 7.59 (1H, dd, J = 8.1, 7.4 Hz); 7.33 (1H, bs); 4.09 (2H, qd, J = 12.9, 7.3 Hz); 3.93 (bs, 2H); 1.92 (1H, hept, J = 6.2 Hz); 1.40–1.25 (8H, m); 0.91 (3H, t, J = 7.4 Hz); 0.87 (3H, t, J = 7.1 Hz). 13C NMR (Chloroform-d, 126 MHz) δ ppm: 164.92, 164.72, 144.93, 133.49, 131.77, 127.78, 127.40, 123.85, 122.89, 122.66, 122.32, 114.45, 44.27, 38.06, 30.88, 28.85, 24.18, 23.22, 14.24, 10.79. FT-IR νmax 2931, 1652, 1598, 1346, 1328, 1300, 787 cm−1. Anal. calcd. C20H24N2O2: C, 74.05; H, 7.46; N, 8.63; found: C, 74.18; H, 7.33; N, 8.79. HRMS (ESI) m/z 325.1916 (calcd for C20H25N2O2 [M + H]+ 325.1911).
3.5. Synthesis of N-(2-Ethylhexyl)-3-amino-4-bromo-1,8-naphthalimide 5
To a solution of N-(2-ethylhexyl)-3-amino-1,8-naphthalimide 4 (100.0 mmol, 32.44 g) in 300 mL acetic acid, a solution of bromine (120.0 mmol, 19.20 g) in 50 mL of acetic acid was added dropwise for five minutes at room temperature. The mixture was stirred for an additional 15 min and then poured into ice (500 g). The precipitate was filtered, washed with water, and dried. The crude product was dissolved in dichloromethane (200 mL), filtered through silica (50 g), washed with dichloromethane, and evaporated to dryness. The solid was dissolved again in a minimum amount of dichloromethane and precipitated with hexane. The precipitate was filtered, washed with hexane, and dried. Yield 39.93 g (99%) as yellow crystals, mp: 177–179 °C. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 8.35 (1H, d, J = 7.3 Hz); 8.31 (1H, d, J = 8.5 Hz), 8.07 (1H, s), 7.71 (1H, dd, J = 8.4, 7.4 Hz), 4.69 (2H, s), 4.08 (2H, qd, J = 12.9, 7.3 Hz), 1.91 (1H, hept, J = 6.3 Hz), 1.42–1.25 (8H, m), 0.92 (3H, t, J = 7.4 Hz), 0.87 (3H, t, J = 7.1 Hz). 13C NMR (Chloroform-d, 126 MHz) δ ppm: 164.49, 164.24, 143.37, 132.01, 131.00, 128.47, 127.86, 123.05, 122.92, 122.77, 121.67, 110.16, 44.35, 38.04, 30.87, 28.84, 24.19, 23.21, 14.24, 10.79. FT-IR νmax 3358, 1650, 1617, 1599, 1231, 780 cm−1. Anal. calcd. C20H23BrN2O2: C, 59.56; H, 5.75; N, 6.95; found: C, 59.79; H, 5.91; N, 7.19. HRMS (ESI) m/z 403.1025 (calcd for C20H24BrN2O2 [M + H]+ 403.1016).
3.6. Synthesis of N-(2-Ethylhexyl)-3-amino-4-chloro-1,8-naphthalimide 6
To a solution of N-(2-ethylhexyl)-3-amino-1,8-naphthalimide 4 (100.0 mmol, 32.44 g) in 150 mL of DMF, N-chlorosuccinimide (120.0 mmol, 16.02 g) was added for one hour at room temperature. The mixture was stirred for an additional four hours and then poured into ice (300 g). The precipitate was filtered, washed with water, and dried. The crude product was dissolved in dichloromethane (200 mL), filtered through silica (50 g), washed with dichloromethane, and evaporated to dryness. The solid was dissolved again in a minimum amount of dichloromethane and precipitated with hexane. The precipitate was filtered, washed with hexane, and dried. Yield 35.17 g (98%) as yellow crystals, mp: 171–172 °C. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 8.35 (1H, dd, J = 7.3, 1.0 Hz); 8.31 (1H, dd, J = 8.5, 1.0 Hz); 8.09 (1H, s); 7.71 (1H, dd, J = 8.5, 7.3 Hz); 4.46 (2H, bs); 4.08 (2H, qd, J = 12.9, 7.3 Hz); 1.91 (1H, hept, J = 6.2 Hz); 1.41–1.25 (8H, m); 0.92 (3H, t, J = 7.5 Hz); 0.87 (3H, t, J = 7.1 Hz). 13C NMR (Chloroform-d, 126 MHz) δ ppm: 164.57, 164.18, 141.82, 130.56, 128.46, 128.21, 127.87, 123.00, 122.92, 122.01, 121.88, 117.65, 44.34, 38.04, 30.87, 28.84, 24.18, 23.21, 14.23, 10.78. FT-IR νmax 1650, 1619, 1427, 1231, 1185, 780 cm−1. Anal. calcd. C20H23ClN2O2: C, 66.94; H, 6.46; N, 7.81; found: C, 66.72; H, 6.65; N, 8.02. HRMS (ESI) m/z 359.1528 (calcd for C20H24ClN2O2 [M + H]+ 359.1521).
General procedure for the preparation of imides 7 and 8: To a cold (0–5 °C) suspension of imide 5 or 6 (40.0 mmol) in 200 mL acetic acid, 100 mL of acetonitrile and 30 mL of 70% sulfuric acid, a solution of sodium nitrite (50.0 mmol, 3.45 g) in 20 mL of water was added dropwise for one hour. The mixture was stirred for an additional 15 min and then poured into the solution of copper (I) bromide (50.0 mmol, 7.17 g) in 80 mL of 48% hydrobromic acid. The mixture was diluted with water up to 600 mL and stirred for two hours. The mixture was extracted with dichloromethane, dried, and evaporated to dryness. The crude product was purified by column chromatography on silica using hexane/dichloromethane as eluent.
3.7. Synthesis of N-(2-Ethylhexyl)-3,4-dibromo-1,8-naphthalimide 7
Yield 16.63 g (89%) as a white solid, mp: 128.5–130.3 °C. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 8.72 (1H, s), 8.63 (1H, d, J = 7.3 Hz), 8.61 (1H, d, J = 8.6 Hz), 7.84 (1H, dd, J = 8.2, 7.7 Hz), 4.10 (2H, qd, J = 12.9, 7.3 Hz), 1.91 (1H, hept, J = 6.1 Hz), 1.41–1.26 (8H, m), 0.92 (3H, t, J = 7.4 Hz), 0.88 (3H, t, J = 7.0 Hz). 13C NMR (Chloroform-d, 126 MHz,) δ ppm: 163.77, 163.17, 135.03, 134.14, 132.49, 131.99, 131.91, 129.25, 127.46, 125.49, 123.41, 123.01, 44.53, 38.03, 30.85, 28.81, 24.17, 23.19, 14.23, 10.75. FT-IR νmax 1702, 1652, 1346, 1221, 1186, 722 cm−1. Anal. calcd. C20H21Br2NO2: C, 51.42; H, 4.53; N, 3.00; found: C, 51.25; H, 4.49; N, 3.11. HRMS (ESI) m/z 466.0007 (calcd for C20H22Br2NO2 [M + H]+ 466.0012).
3.8. Synthesis of N-(2-Ethylhexyl)-3-bromo-4-chloro-1,8-naphthalimide 8
Yield 12.34 g (73%) as an off-white solid, mp: 123–124 °C. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 8.72 (1H, s); 8.63 (1H, dd, J = 7.3, 1.0 Hz); 8.59 (1H, dd, J = 8.5, 1.0 Hz); 7.86 (1H, dd, J = 8.5, 7.3 Hz); 4.10 (2H, qd, J = 12.9, 7.3 Hz); 1.91 (1H, hept, J = 6.2 Hz); 1.44–1.21 (8H, m); 0.92 (3H, t, J = 7.4 Hz); 0.87 (3H, t, J = 7.0 Hz). 13C NMR (Chloroform-d, 126 MHz) δ ppm: 163.83, 163.03, 138.45, 135.15, 131.95, 131.06, 130.68, 128.97, 127.72, 123.42, 122.43, 122.33, 44.52, 38.03, 30.85, 28.81, 24.17, 23.19, 14.23, 10.75. FT-IR νmax 1704, 1654, 1590, 1347, 1224, 783 cm−1. Anal. calcd. C20H21BrClNO2: C, 56.82; H, 5.01; N, 3.31; found: C, 56.99; H, 5.06; N, 3.15. HRMS (ESI) m/z 422.0515 (calcd for C20H22BrClNO2 [M + H]+ 422.0517).
General procedure for the preparation of imides 9 and 10: To a cold (0–5 °C) suspension of imide 5 or 6 (40.0 mmol) in 200 mL acetic acid, 100 mL of acetonitrile and 30 mL of 70% sulfuric acid, a solution of sodium nitrite (50.0 mmol, 3.45 g) in 20 mL of water was added dropwise for one hour. The mixture was stirred for an additional 15 min and then was poured into the solution of copper (I) chloride (50.0 mmol, 4.95 g) in 80 mL of 37% hydrochloric acid. The mixture was diluted with water up to 600 mL and stirred for two hours. The mixture was extracted with dichloromethane, dried, and evaporated to dryness. The crude product was purified by column chromatography on silica using hexane/dichloromethane as eluent.
3.9. Synthesis of N-(2-Ethylhexyl)-4-bromo-3-chloro-1,8-naphthalimide 9
Yield 12.01 g (71%) as an off-white solid, mp: 127–129 °C. 1H NMR (Chloroform-d, 500 MHz,) δ ppm: 8.62 (1H, dd, J = 7.3, 1.1 Hz); 8.58 (1H, dd, J = 8.2, 1.1 Hz); 8.57 (1H, s); 7.86 (1H, dd, J = 8.5, 7.3 Hz), 4.10 (2H, qd, J = 12.9, 7.3 Hz), 1.91 (1H, hept, J = 6.2 Hz), 1.46–1.21 (8H, m), 0.92 (3H, t, J = 7.4 Hz), 0.87 (3H, t, J = 7.1 Hz). 13C NMR (Chloroform-d, 126 MHz) δ ppm: 163.79, 163.23, 135.01, 133.67, 132.32, 132.03, 131.84, 129.42, 129.21, 127.15, 123.33, 123.10, 44.53, 38.03, 30.85, 28.80, 24.17, 23.19, 14.23, 10.75. FT-IR νmax 1703, 1652, 1588, 1346, 1224, 1186, 783 cm−1. Anal. calcd. C20H21BrClNO2: C, 56.82; H, 5.01; N, 3.31; found: C, 56.66; H, 4.93; N, 3.49. HRMS (ESI) m/z 422.0512 (calcd for C20H22BrClNO2 [M + H]+ 422.0517).
3.10. Synthesis of N-(2-Ethylhexyl)-3,4-dichloro-1,8-naphthalimide 10
Yield 10.44 g (69%) as a white solid, mp: 124–125 °C. 1H NMR (Chloroform-d, 500 MHz) δ ppm: 8.62 (1H, dd, J = 7.3, 1.0 Hz); 8.58 (1H, s); 8.54 (1H, dd, J = 8.6, 1.0 Hz); 7.87 (1H, dd, J = 8.5, 7.3 Hz); 4.10 (2H, qd, J = 12.9, 7.3 Hz); 1.91 (1H, hept, J = 6.2 Hz); 1.41–1.26 (8H, m); 0.92 (3H, t, J = 7.4 Hz); 0.87 (3H, t, J = 7.0 Hz). 13C NMR (Chloroform-d, 126 MHz) δ ppm: 163.86, 163.11, 136.29, 132.48, 132.23, 131.86, 130.74, 130.65, 128.96, 127.33, 123.35, 122.38, 44.52, 38.03, 30.84, 28.80, 24.17, 23.19, 14.23, 10.75. FT-IR νmax 1704, 1653, 1347, 1312, 1187, 783 cm−1. Anal. calcd. C20H21Cl2NO2: C, 63.50; H, 5.60; N, 3.70; found: C, 63.81; H, 5.39; N, 3.47. HRMS (ESI) m/z 378.1019 (calcd for C20H22Cl2NO2 [M + H]+ 378.1022).
4. Conclusions
A reliable protocol for a multigram-scale synthesis of 3,4-dihalogeno-1,8-naphthalimides is proposed. The target compounds are obtained easily from commercially available and cheap 1,8-naphthalic anhydride. The strategy allows the preparation of 3,4-dibromo, 3,4-dichloro, or mixed (3-bromo-4-chloro or 4-bromo-3-chloro) NI derivatives. Due to their specific functionalization, we believe the proposed imides are powerful building-block molecules in rylene chemistry, as they can be further derivatized by direct aromatic nucleophilic substitution or metal-catalyzed cross-coupling reactions. All reaction steps proceed on a gram scale and in very high yields. The purification of the intermediate and final compounds is easy and straightforward compared to the use of anhydrides due to imidization in the early stage.
Supplementary Materials
1H-, 13C-NMR, IR and HRMS spectra copies of synthesized compounds can be found in the File Supplementary Materials.
Author Contributions
Conceptualization, M.M. and Y.Z.; investigation, M.M., D.A., N.S., I.Z. and Z.V.; writing—original draft preparation, Y.Z. and S.S.; writing—review and editing, S.S. and Y.Z.; visualization, M.M. and I.Z.; supervision, Y.Z.; project administration, Y.Z.; funding acquisition, Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Bulgarian National Science Fund, grant number KP-06-H61/1.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Acknowledgments
Y.Z., S.S., M.M. gratefully acknowledge the funding by the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project No BG-RRP-2.004-0008.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gong, H.-H.; Addla, D.; Lv, J.-S.; Zhou, C.-H. Heterocyclic Naphthalimides as New Skeleton Structure of Compounds with Increasingly Expanding Relational Medicinal Applications. Curr. Top. Med. Chem. 2016, 16, 3303–3364. [Google Scholar] [CrossRef]
- Poddar, M.; Sivakumar, G.; Misra, R. Donor–Acceptor Substituted 1,8-Naphthalimides: Design, Synthesis, and Structure–Property Relationship. J. Mater. Chem. C 2019, 7, 14798–14815. [Google Scholar] [CrossRef]
- Jain, N.; Kaur, N. A Comprehensive Compendium of Literature of 1,8-Naphthalimide Based Chemosensors from 2017 to 2021. Coord. Chem. Rev. 2022, 459, 214454. [Google Scholar] [CrossRef]
- Geraghty, C.; Wynne, C.; Elmes, R.B. 1,8-Naphthalimide Based Fluorescent Sensors for Enzymes. Coord. Chem. Rev. 2021, 437, 213713. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, C.; Su, M.; Rong, X.; Zhang, Y.; Wang, X.; Wang, K.; Li, X.; Yu, Y.; Zhang, X.; et al. Recent Advances in 4-Hydroxy-1,8-Naphthalimide-Based Small-Molecule Fluorescent Probes. Coord. Chem. Rev. 2021, 448, 214153. [Google Scholar] [CrossRef]
- Nie, W.; Hu, L. Design of 1,8-Naphthalimide-Based Fluorescent Functional Molecules for Biological Application: A Review. ChemistrySelect 2024, 9, e202303779. [Google Scholar] [CrossRef]
- Tandon, R.; Luxami, V.; Tandon, N.; Paul, K. Recent Developments on 1,8-Naphthalimide Moiety as Potential Target for AntiCancer Agents. Bioorg. Chem. 2022, 121, 105677. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.D.; Walczak, K.Z. l,8-Naphthalimide Based DNA Intercalators and Anticancer Agents. A Systematic Review from 2007 to 2017. Eur. J. Med. Chem. 2018, 159, 393–422. [Google Scholar] [CrossRef] [PubMed]
- Kagatikar, S.; Sunil, D. A Systematic Review on 1,8-Naphthalimide Derivatives as Emissive Materials in Organic Light-Emitting Diodes. J. Mater. Sci. 2022, 57, 105–139. [Google Scholar] [CrossRef]
- Yu, H.; Guo, Y.; Zhu, W.; Havener, K.; Zheng, X. Recent Advances in 1,8-Naphthalimide-Bbased Small-Molecule Fluorescent Probes for Organelles Imaging and Tracking in Living Cells. Coord. Chem. Rev. 2021, 444, 214019. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Zagranyarski, Y.; Müller, C.; Schulz, M.; Kupfer, S.; Görls, H.; El-Khateeb, M.; Gräfe, S.; Dietzek, B.; Peneva, K.; et al. [FeFe]-Hydrogenase H-cluster mimics mediated by naphthalene monoimide derivatives of peri-substituted dichalcogenides. Dalton Trans. 2017, 46, 11180–11191. [Google Scholar] [CrossRef] [PubMed]
- Mutovska, M.; Skabeev, A.; Konstantinov, K.; Cabanetos, C.; Stoyanov, S.; Zagranyarski, Y. One-pot Synthesis of Fused-Rings Heterocyclic Systems Based on Symmetrically Benzofuran Annulated 1,8-Naphthalimides. Dye. Pigment. 2023, 220, 111701. [Google Scholar] [CrossRef]
- Zagranyarski, Y.; Mutovska, M.; Petrova, P.; Tomova, R.; Ivanov, P.; Stoyanov, S. Dioxin-Annulated 1,8-Naphthalimides—SynThesis, Spectral and Electrochemical Properties, and Application in OLED. Dye. Pigment. 2021, 184, 108585. [Google Scholar] [CrossRef]
- Braña, M.F.; Morán, M.; de Vega, M.J.P.; Pita-Romero, I.; Walker, N. Synthesis and Cytostatic Activity of Enynes, Enediynes and Dienediynes Linked to Intercalators. Tetrahedron 1995, 51, 9127–9138. [Google Scholar] [CrossRef]
- Meng, X.; Zhu, W.; Zhang, Q.; Feng, Y.; Tan, W.; Tian, H. Novel Bisthienylethenes Containing Naphthalimide as the Center Ethene Bridge: Photochromism and Solvatochromism for Combined NOR and INHIBIT Logic Gates. J. Phys. Chem. B 2008, 112, 15636–15645. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Song, L.; Yang, Y.; Tian, H. Novel Bisthienylethene Containing Ferrocenyl-Substituted Naphthalimide: A Photo- and Redox Multi-Addressable Molecular Switch. Chem.-A Eur. J. 2012, 18, 13388–13394. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ge, H.; Chen, Z.; Liang, J.; Huang, J.; Zhang, Y.; Chen, X.; Meng, X.; Liu, S.H.; Yin, J. Imides Modified Benzopicenes: Synthesis, Solid Structure and Optoelectronic Properties. Org. Biomol. Chem. 2014, 12, 8902–8910. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chen, Z.; Zhang, Y.; Zhang, J.; Liu, S.H.; Yin, J. Imide-Modified Dinaphtho[1,2-b:2′,1′-d]Thiophene and Dinaphtho[1,2-b:2′,1′-d]Thiophene 13,13-Dioxide: Synthesis and Optoelectronic Properties. J. Org. Chem. 2015, 80, 8443–8448. [Google Scholar] [CrossRef] [PubMed]
- Kulik, L.V.; Krivenko, O.L.; Nevostruev, D.A.; Kobeleva, E.S.; Kravets, N.V.; Uvarov, M.N.; Molchanov, I.A.; Dmitriev, A.A.; Kazantsev, M.S.; Gatilov, Y.V.; et al. Aryl-Bridged Thienonaphthalimides: Synthesis, Characterization and Optoelectronic Properties. Eur. J. Org. Chem. 2024, 27, e202300848. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).